Fungal culture and plant materials
The Foc fungal strain was isolated from the experimental field of Yangzhou University, Jiangsu Province, China, and propagated on PDA plates at 28 ℃ for 4 days, then cultured in potato dextrose broth on a shaker at 180 rpm at 28 ℃ for 3 days. The spore suspension was diluted to 1 × 106 spores per milliliter with sterile distilled water.
Seedlings of cucumber ‘Rijiecheng’ and ‘Superina’, moderately Foc-resistant cultivar and Foc-sensitive cultivar, were grown in 32-well plates filled with an aseptic organic substrate (contents of total nitrogen, phosphorus, and potassium = 40-60 g/kg, content of humus ≥ 350 g/kg, pH = 6.5-7.5) at 25 ℃/18 ℃ day/night temperatures with a 16 h/8 h photoperiod. Seedlings were infected with Foc by irrigation of the roots with a fungal spore suspension (5 mL per seedling) at the second-true-leaf stage. Cucumber roots were sampled at 0, 24, 48, 96, and 192 h after inoculation with three biological replicates. All the seeds were obtained from the homozygous inbred lines of our own laboratory.
RNA extraction, cDNA library construction, Illumina sequencing, and analysis of sequence reads
Total RNA was isolated using the TaKaRa MiniBEST Plant RNA Extraction Kit (TaKaRa, China). The RNA concentration was measured using the Qubit RNA Assay Kit with a Qubit® 2.0 fluorometer (Life Technologies, USA). One microgram of RNA was used as input for the RNA sample preparations. The mRNA was purified from the total RNA using poly-T oligo-attached magnetic beads. Fragmentation was carried out using divalent cations under an elevated temperature in NEBNext® First Strand Synthesis Reaction Buffer (5×). The first-strand cDNA was synthesized using the mRNA fragments as templates. To select cDNA fragments of preferentially 200–250 bp in length, the library fragments were purified with the AMPure XP system (Beckman Coulter, USA). Eligible cDNAs were selected for PCR amplification, which was performed with Phusion® High-Fidelity DNA polymerase, Universal PCR primers, and Index (X) Primer. The PCR products were purified using the AMPure XP system and library quality was assessed with an Agilent Bioanalyzer 2100 system.
Primary cDNA produced using the Illumina HiSeq 2500 platform by BioMarker Technologies (Beijing, China) were termed raw reads. Clean reads were obtained by removing reads containing the adapter, reads containing poly-N, and low-quality reads. In addition, Q20, Q30, GC-content and sequence duplication values of the clean reads were calculated. The trimmed reads were aligned to the cucumber Chinese Long reference genome v2 [47], which was retrieved from http://cucurbitgenomics.org/organism/2. All downstream analyses were based on the clean, high-quality data. Quantification of gene expression levels was estimated as fragments per kilobase of transcript per million fragments mapped (FPKM) [48, 49] using Cufflinks (version: 2.1.1).
Identification of DEGs and validation of RNA-seq by qPCR
We divided the data into four groups by comparing the data at 0 h with that at the other sampling time points. The analysis of DEGs for the four groups was performed using the DESeq R package (1.10.1). DESeq provides statistical routines for determining differential expression in digital gene expression data using a model based on the negative binomial distribution. The P values were adjusted using the Benjamini–Hochberg approach for controlling the FDR. Genes with an adjusted FDR < 0.01 identified by DESeq and log2 FPKM (fold change) ≥ 1 were considered to be differentially expressed.
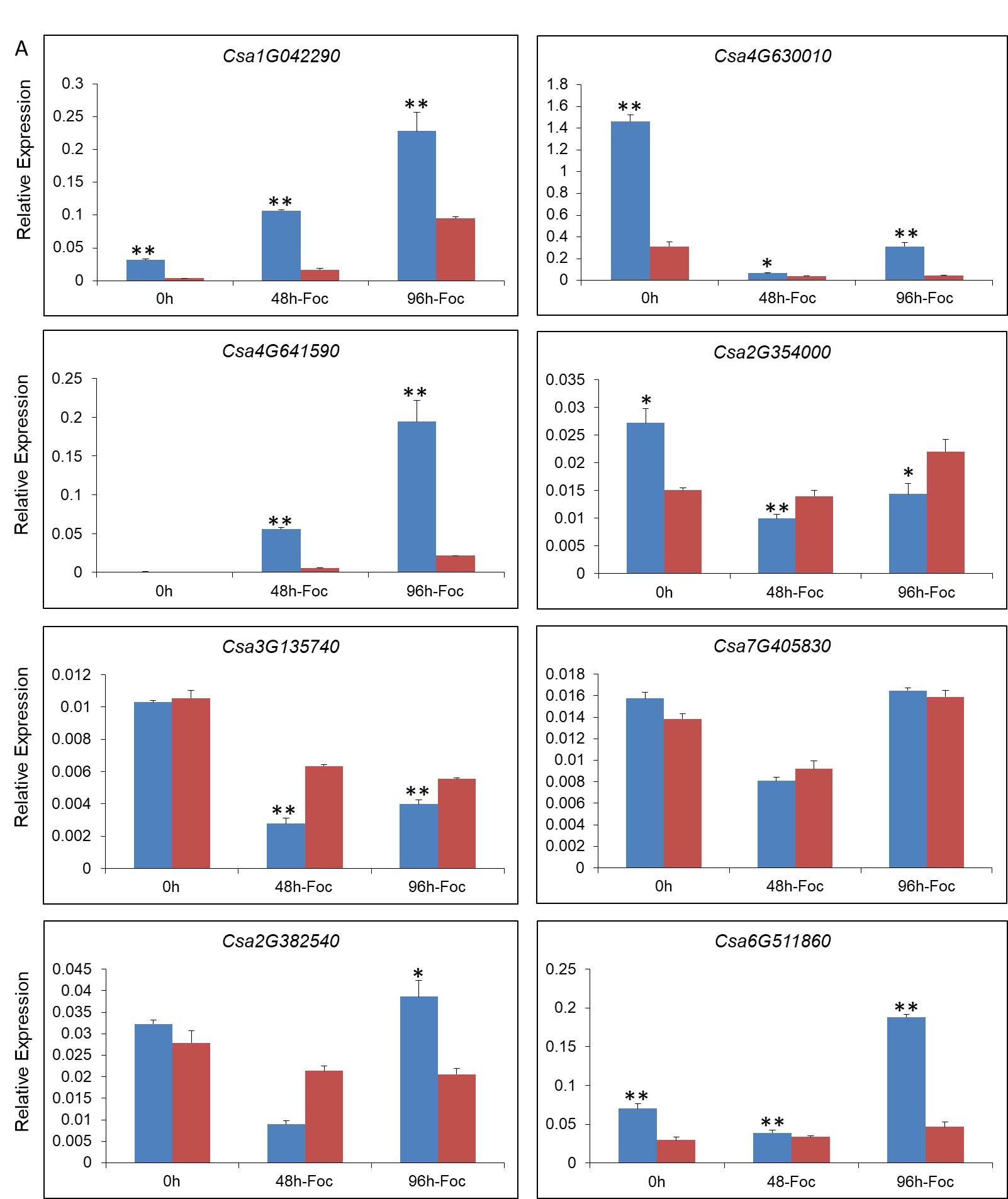
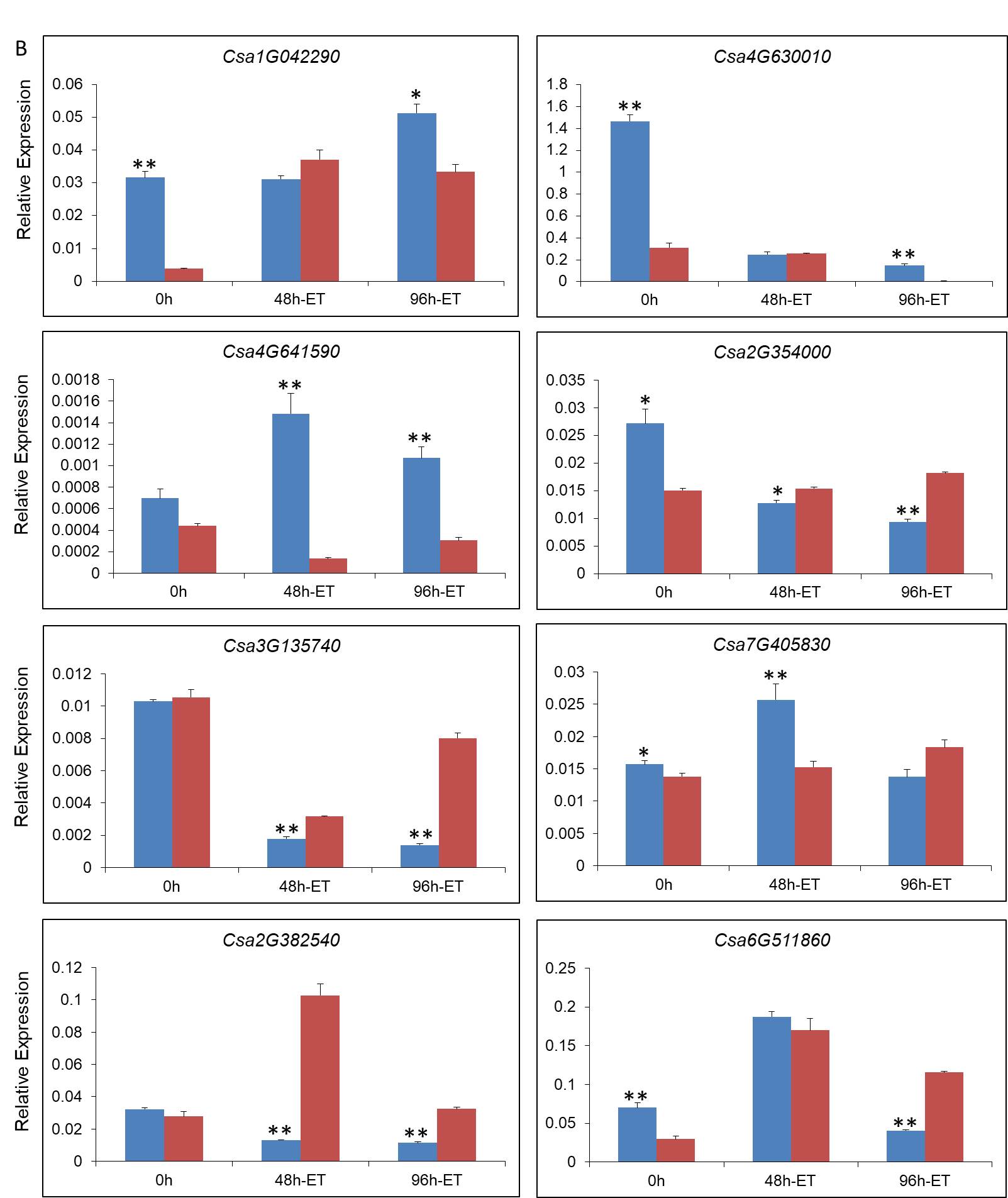
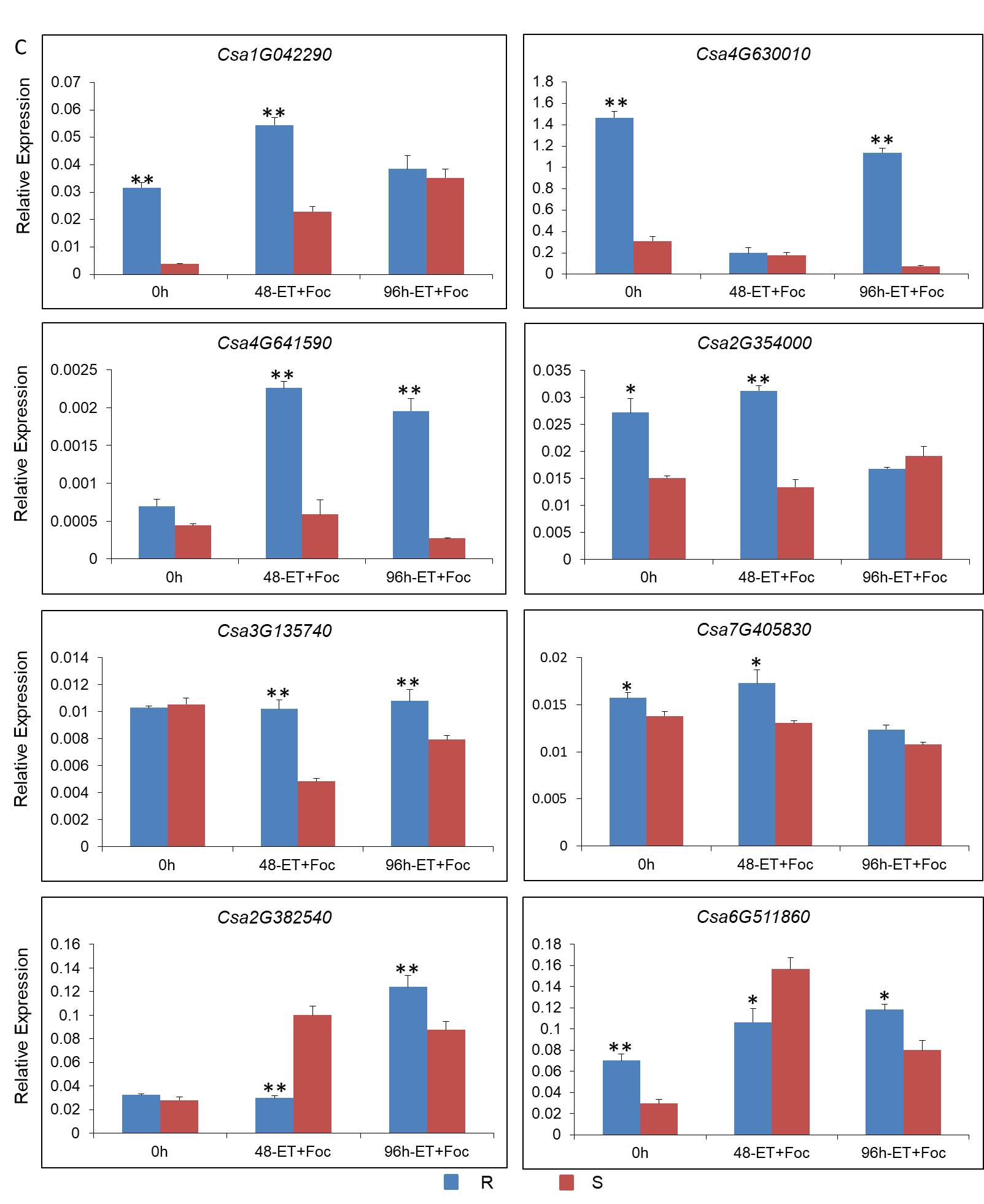
The root portion was placed as the expression samples. Total RNA of each condition was isolated using the MiniBEST Plant RNA Extraction Kit (TaKaRa, China), then dissolved with UltraPure™ DNase/RNase-free Distilled Water (Invitrogen, USA). Total RNA was reverse transcribed using the PrimeScript™ RT Reagent Kit with gDNA Eraser (TaKaRa, China). Primer sequences for qPCR were designed using Primer Premier 5. The qPCR analysis was performed using SYBR® Premix Ex Taq™ Ⅱ (TaKaRa, China) in accordance with the manufacturer’s instructions. SYBR Green PCR cycling was performed on an Iqtm5 Multicolor qPCR detection system (Bio-Rad, USA) using 20 μL samples with the following temperature program: 95 ℃ for 3 min, followed by 39 cycles of 95 ℃ for 10 s, 60 ℃ for 20 s, and 72 ℃ for 20 s, then a melting curve analysis was performed. The relative quantification of gene expression was calculated and normalized to the tubulin alpha chain gene (Csa4G000580). Three biological replicates from each condition were used for qPCR.
Evaluation of exogenous ethephon on Foc and cucumber seedlings
To ascertain the effect of ET on Foc growth, exogenous ethephon (a donor of ET) was incorporated in PDA medium. To 18 mL PDA medium was added either 2 mL ethephon (final concentration 1000 ppm) or 2 mL sterile water (as the control). Foc was cultured on the PDA medium at 25 ℃ for 5 days and the colony diameter was measured three times.
To examine the effect of ET on seedling resistance to Foc, one third of seedlings roots treated with mock solutions as the control, one third were inoculated with a Foc strain (spore concentration 106 conidia/mL) using the root irrigation method, and the others were inoculated with Foc and sprayed with exogenous ethephon solution (concentration 10 ppm) on leaves at the same time. After three weeks, disease grades were recorded. The disease grades were as follows: Grade 0, asymptomatic; Grade 1, slightly discolored of stem base and cotyledon; Grade 2, necrotic patches at the stem base and slight wilting of the seedling; Grade 3, color of necrotic patches at the stem kept deep with longitudinal fissure and the seedling was visibly wilting; Grade 4, the seedling was dead. The disease index was calculated using the following formula: Disease index (%) = ×100. Each experimental group included at least 15 individual seedlings and three biological replicates were treated. Mock inoculated seedlings were used as the control.
Measurement of endogenous ET biosynthesis
The two cucumber inbred lines, ‘Rijiecheng’ and ‘Superina’, which are resistant and sensitive to Foc, respectively, were used in this experiment. Seedlings at the second-true-leaf stage were inoculated by irrigation with Foc (spore concentration 106 conidia/mL) and sprayed with exogenous ethephon solution (concentration 10 ppm). Endogenous ET biosynthesis by the root portion of the seedlings was measured at 48 and 96 h after treatment. The root portion was placed in a sealed 30 mL ampoule for 24 h in the dark. For analysis of ET production, gas samples (1 mL) were collected using a syringe and injected into a gas chromatograph (Agilent 7890A, USA) fitted with a flame ionization and electron capture detectors [50]. Experiments were conducted with three biological replicates.
{kind=link}
{kind=link}
{kind=link}