Culture-independent methods have led to a deeper appreciation of the microbes associated with the human gut (Stewart, 2018), plant rhizosphere (Berendsen et al., 2012), and various open or natural environments (Thompson et al., 2017). The interpretation of 16S rRNA gene sequence data relies on determination of true sequence variants through the use of de-noising algorithms followed by assignment to known taxonomic groupings by comparison with sequences in curated databases (Bokulich et al., 2018). Assemblages of eubacterial and archaeal organisms (Parada et al., 2016) in the phycosphere of experimental, outdoor open algal production reactors (DOE-RAFT project DE-EE0006269, The University of Arizona) were identified by comparative analysis of the 16S rRNA gene sequence. Environmental and experimental variables were collected over more than two years of cultivation and correlated with shifts in the bacterial community structure. The associations between bacteria and microalgal biomass productivity and of culture ‘health’ under a scenario of repeated rounds of infection of the algal host by the predatory bacterial pathogen, V. chlorellavorous, were investigated to help identify conditions to guide optimization of biomass production in the RAFT reactors. Results demonstrated that the community was dynamic, complex, and responsive to several key environmental variables. While the approaches used in this study were able to capture evidence of a fluctuating eubacterial composition associated with the C. sorokiniana algal cultures, the specific mechanisms guiding the observed biological fluxes are not yet known. However, it was possible to apply available information about certain closest bacterial relatives obtained from previously studied phycosphere and rhizosphere systems to develop hypotheses for further testing.
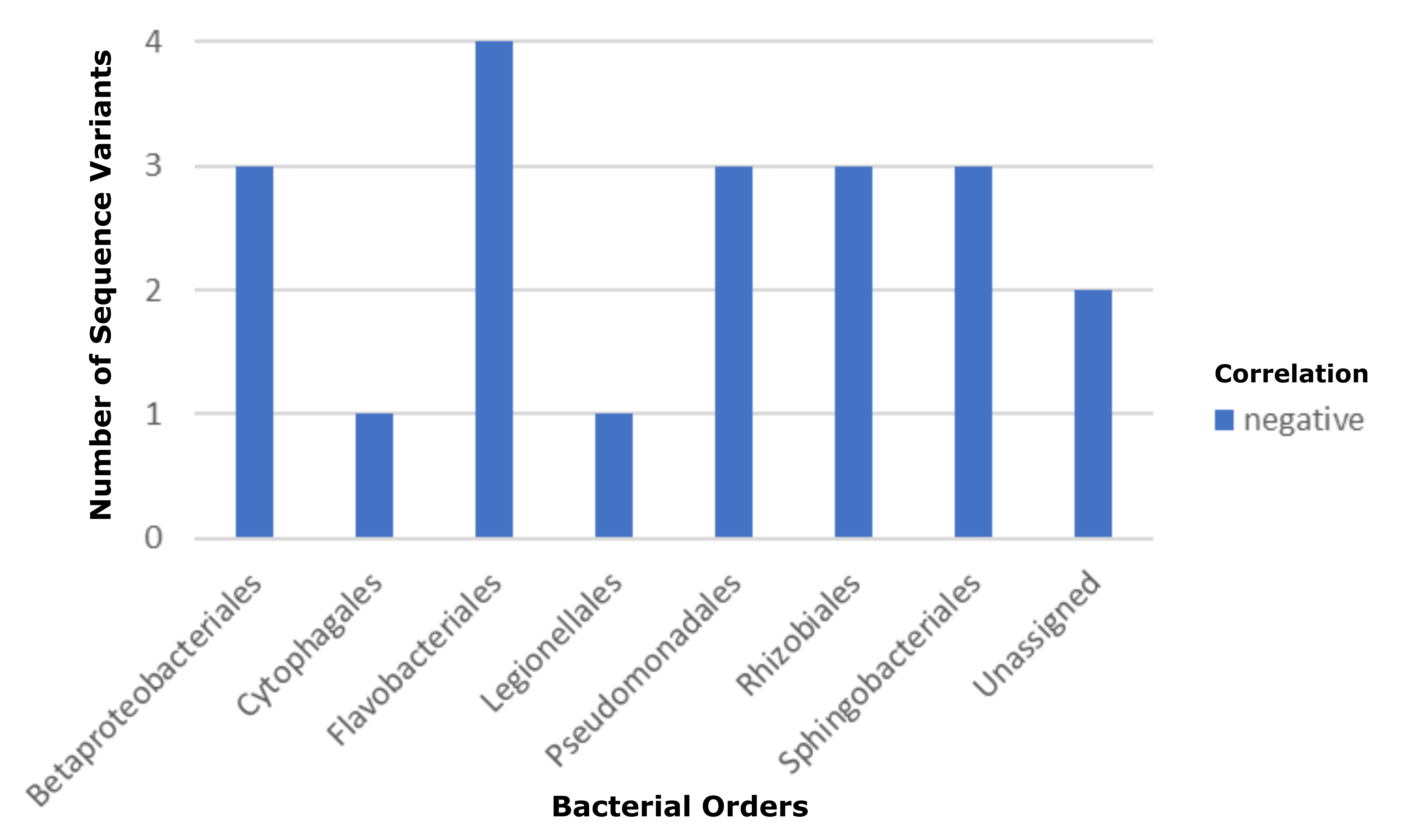
The bacterial phycosphere composition changed throughout the different cultivation cycles and over time, with community diversity increasing (Fig. 2), while certain taxa became more or less prevalent than others (Fig. 3). The fixation of gaseous nitrogen into bioavailable ammonium in exchange for organic carbon is one of the most well-known reactions involving land plants and rhizosphere-associated microorganisms (Berendsen et al., 2012). An analogous phycosphere association has been reported to occur in marine environments where widely-distributed prokaryotes that are associated with their unicellular phytoplankton express the nifH nitrogenase gene, among other dynamic mutualistic contributions (Thompson et al., 2012). The order Rhizobiales, which contains numerous nitrogen-fixing rhizosphere symbionts, was overrepresented among ASVs significantly associated with algal biomass productivity (Fig. 6). Within the Rhizobiales, bacteria belonging to the genus, Devosia, represented two of the ten ASVs whose presence was most closely-associated with C. sorokiniana growth (Table 2). The genus, Devosia contains members that have been previously isolated from root nodules of aquatic leguminous plants (Rivas et al., 2002), suggesting the possibility of a mutualistic partnership with C. sorokiniana involving the partitioning of nitrogen for uptake by algae and other phycosphere members. The Family, Burkholderiaceae sequence variants were very highly correlated with biomass productivity, and contain known nitrogen fixing bacteria (Sawada et al., 2003). The abundance of members of the order, Chitinophagales increased after the initial algal growth phase. This pattern was observed in the baseline experiments for samples collected six-days post-inoculation of reactors (Figs. 3 and 4) and Chitinophagales ASVs were positively correlated with number of days in culture during the seasonal monitoring (Fig. 7). The latter group are related to bacteria capable of consuming chitin, and may be opportunists on other phycosphere bacterial species, or the microalgal cell walls that are composed of chitin and chitosan (Blanc et al., 2010).
Table 2
Most significant sequence variants to biomass productivity according to multinomial model
| Differential | Kingdom | Phylum | Class | Order | Family | Genus | Species |
| 5.81 | Bacteria | Proteobacteria | Alphaproteobacteria | Rickettsiales | SM2D12 | unidentified marine bacterioplankton | unidentified marine bacterioplankton |
| 5.47 | Bacteria | Proteobacteria | Deltaproteobacteria | Bdellovibrionales | Bdellovibrionaceae | Bdellovibrio | Bdellovibrio bacteriovorus |
| 2.86 | Bacteria | Proteobacteria | Gammaproteobacteria | Betaproteobacteriales | Burkholderiaceae | Unassigned | Unassigned |
| 2.84 | Bacteria | Proteobacteria | Gammaproteobacteria | Betaproteobacteriales | Burkholderiaceae | Unassigned | Unassigned |
| 2.72 | Bacteria | Bacteroidetes | Bacteroidia | Cytophagales | Cyclobacteriaceae | Algoriphagus | Algoriphagus sp. ZH062 |
| 2.72 | Bacteria | Proteobacteria | Alphaproteobacteria | Rhizobiales | Devosiaceae | Devosia | Unassigned |
| 2.72 | Bacteria | Proteobacteria | Alphaproteobacteria | Rhizobiales | Devosiaceae | Devosia | Unassigned |
| 2.72 | Bacteria | Proteobacteria | Alphaproteobacteria | Rhizobiales | Xanthobacteraceae | Pseudoxanthobacter | uncultured alpha proteobacterium |
| 2.72 | Bacteria | Bacteroidetes | Bacteroidia | Cytophagales | Spirosomaceae | Unassigned | Unassigned |
| 2.72 | Bacteria | Proteobacteria | Gammaproteobacteria | Betaproteobacteriales | Burkholderiaceae | Curvibacter | Unassigned |
| 2.72 | Bacteria | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | Defluviimonas | Unassigned |
| 2.72 | Bacteria | Proteobacteria | Alphaproteobacteria | Sphingomonadales | Sphingomonadaceae | Unassigned | Unassigned |
| 2.72 | Bacteria | Bacteroidetes | Bacteroidia | Chitinophagales | Chitinophagaceae | Flavihumibacter | uncultured bacterium |
| 2.72 | Bacteria | Planctomycetes | Planctomycetacia | Pirellulales | Pirellulaceae | Rhodopirellula | Unassigned |
| 2.72 | Bacteria | Proteobacteria | Gammaproteobacteria | Betaproteobacteriales | Burkholderiaceae | Hydrogenophaga | Unassigned |
| 2.72 | Unassigned | Unassigned | Unassigned | Unassigned | Unassigned | Unassigned | Unassigned |
| 2.72 | Bacteria | Proteobacteria | Alphaproteobacteria | Rhizobiales | Hyphomicrobiaceae | Hyphomicrobium | Unassigned |
| 2.72 | Bacteria | Acidobacteria | Blastocatellia (Subgroup 4) | Blastocatellales | Blastocatellaceae | uncultured | uncultured bacterium |
| 2.72 | Bacteria | Proteobacteria | Alphaproteobacteria | Rhizobiales | Rhizobiaceae | Unassigned | Unassigned |
| 2.72 | Bacteria | Bacteroidetes | Bacteroidia | Chitinophagales | Chitinophagaceae | Terrimonas | Terrimonas sp. T16R-129 |
| 2.72 | Bacteria | Bacteroidetes | Bacteroidia | Flavobacteriales | Flavobacteriaceae | Flavobacterium | Unassigned |
| 2.72 | Bacteria | Bacteroidetes | Bacteroidia | Chitinophagales | Chitinophagaceae | Parasegetibacter | uncultured bacterium |
| 2.72 | Bacteria | Planctomycetes | Phycisphaerae | Phycisphaerales | Phycisphaeraceae | SM1A02 | metagenome |
| 2.70 | Bacteria | Proteobacteria | Deltaproteobacteria | Oligoflexales | 0319-6G20 | Unassigned | Unassigned |
| 2.70 | Bacteria | Proteobacteria | Deltaproteobacteria | Oligoflexales | Oligoflexaceae | uncultured | uncultured bacterium |
| 2.69 | Bacteria | Proteobacteria | Gammaproteobacteria | Pseudomonadales | Moraxellaceae | Perlucidibaca | uncultured bacterium |
| 2.67 | Bacteria | Proteobacteria | Gammaproteobacteria | Betaproteobacteriales | Burkholderiaceae | Ramlibacter | Unassigned |
| 2.51 | Bacteria | Proteobacteria | Deltaproteobacteria | Bdellovibrionales | Bdellovibrionaceae | Bdellovibrio | Unassigned |
| -2.39 | Bacteria | Proteobacteria | Gammaproteobacteria | Pseudomonadales | Pseudomonadaceae | Pseudomonas | Unassigned |
| -4.65 | Bacteria | Bacteroidetes | Bacteroidia | Cytophagales | Cytophagaceae | Cytophaga | uncultured bacterium |
| -5.40 | Bacteria | Verrucomicrobia | Verrucomicrobiae | Verrucomicrobiales | Rubritaleaceae | Luteolibacter | uncultured bacterium |
| -7.65 | Bacteria | Proteobacteria | Gammaproteobacteria | Oceanospirillales | Pseudohongiellaceae | Pseudohongiella | metagenome |
| -8.03 | Bacteria | Patescibacteria | Parcubacteria | Candidatus Nomurabacteria | Unassigned | Unassigned | Unassigned |
| -8.36 | Bacteria | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | Unassigned | Unassigned |
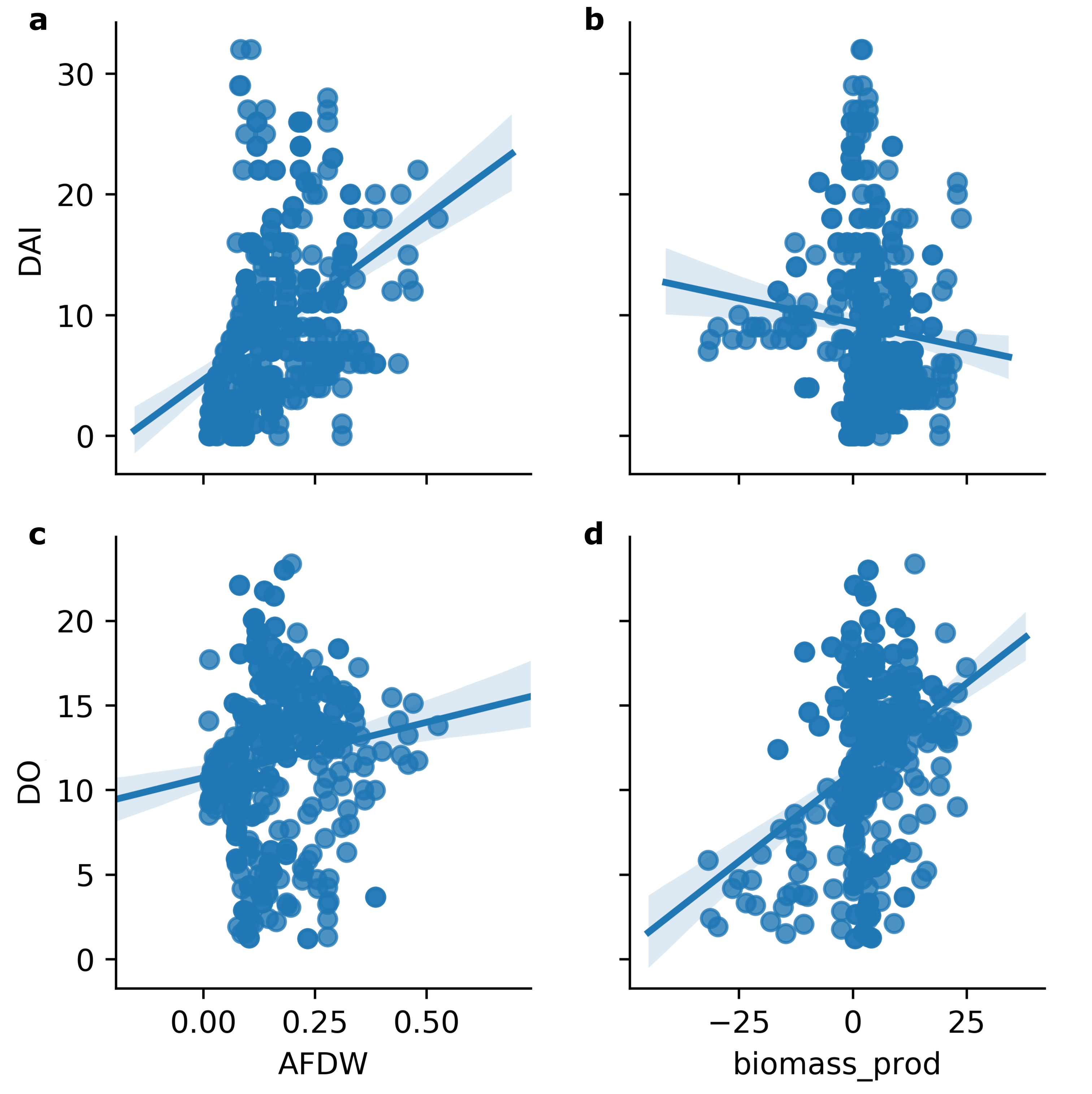
Plant growth promoting rhizobacteria (PGPR) have been shown to be phylogenetically diverse, and illicit increased fitness of their terrestrial plant partners by various mechanisms (Berendsen et al., 2012; Lugtenberg and Kamilova, 2009). Several studies have focused on the promotion of algae growth by taxonomically and functionally diverse bacteria. In one example, Rhizobium spp. were the major phycosphere constituents for four different green microalgal species, including Chlamydomonas reinhardtii P.A. Dangeard 1888, Chlorella vulgaris Beyerinck 1890, Scenedesmus sp., and Botryococcus braunii Kützing (Kim et al., 2014). The most commonly occurring full length 16S rRNA gene sequence in the study was identified as a Rhizobium spp. (EU781656) (Kim et al. 2014). In the RAFT reactors, an isolate sharing 100% nucleotide identity with this Rhizobium spp., ASV (b449f5066387be07ee577a95585d45ca), was present in 39.7% of samples analyzed during routine monitoring studies. Also, a second Rhizobium spp. (JX255399), following growth in pure culture, was shown to promote the growth of the four algal species following inoculation of the algal cultures (Kim et al. 2014 ). This bacterial 16S rRNA sequence was 100% identical to the ASV, 8ce12e88f6b59bb09494567f0d678092, from the RAFT data set for which it was present in 5.6% of the samples. However, neither the ubiquitous nor the growth-promoting Rhizobium spp. were significantly correlated with biomass productivity, based on coefficients of -0.012 and − 0.118, respectively (Additional File 5: Table S2), and, the linear regression analysis identified one insignificant positive relationship (Fig. 9c). These observations indicate that while phylogenetically-identical bacterial species may apparently occur in different phycospheres, their specific contribution to the community may be specific to habitat and/or algal species composition.
In a recent study, several Azospirillum spp. were shown to increase the growth rate of Chlorella sorokiniana (Shihira & R.W.Krauss) facilitated by its contribution the hormone, indole-3-acetic acid (IAA) (De-Bashan et al., 2008). Additional studies showed that algae growth increased following inoculation of cultures with Bacillus pumilus (Amavizca et al., 2017; Bashan et al., 2016; Hernandez et al., 2009). The RAFT samples contained seven ASVs that are assigned to the order Azospirillales, however, they were found to be negatively correlated with biomass productivity. While the sole Bacillus sp. ASV identified in the RAFT samples during monitoring had a -0.093 correlation with biomass productivity, two of the six members of the order Bacillales were positively linked to growth. Further, a Chlorella spp. culture having a low growth rate, has been shown to exhibit increased growth when grown in laboratory cultures to which a bacterial consortium from a fast-growing algal culture was used to augment productivity. An analysis of the relative abundances of 16S rRNA gene sequences revealed that several Ruegeria sp. were enriched in the fast-growing cultures. While no members of the genus, Ruegeria ASVs were identified among the RAFT samples, the order Rhodobacteriales to which Ruegeria is assigned, was highly correlated with biomass productivity and rapid growth in lab cultures (Richter et al., 2018).
Bacteria that were negatively associated with algal growth during the RAFT project may have been antagonistic or possibly pathogenic to C. sorokiniana, and so are prospective candidates of taxa that could serve as indicators for declining algal culture health. Six ASVs were identified that were significantly negatively associated with C. sorokiniana biomass productivity, based on the multinomial model. These were identified as members of the Rhodobacteraceae, order Nomurabacteria, and the genera Pseudohongiella, Luteolibacter, Cytophaga, and Pseudomonas. (Table 2). Among them, only the genus, Cytophaga has been robustly linked to algicidal activity and killing of diatom species (Imai et al., 1993; Mayali and Azam, 2004), predation on cyanobacteria (Rashidan and Bird, 2001), and accumulation during stationary and death phases of a mixed photobioreactor culture containing members of the family, Scenedesmaceae (Carney et al., 2014).
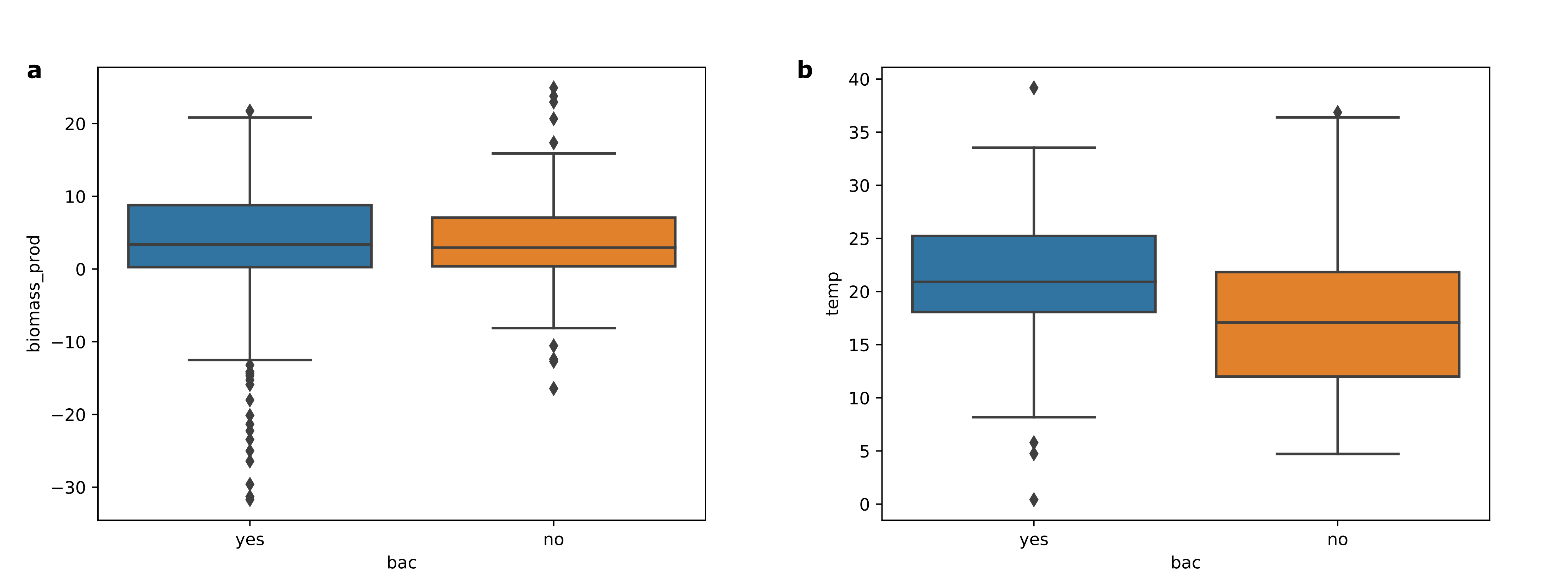
The predatory cyanobacterium, V. chlorellavorus, is recognized as a virulent pathogen of several species of Chlorella (Coder and Goff, 1986; Ganuza et al., 2016b; Soo et al., 2015). It has been previously shown to be responsible for the death of C. sorokiniana cultures in Arizona at the RAFT site (Steichen and Brown, 2018). Experiments carried out in this study showed that the infection cycle could be managed to some extent through applications of the quaternary ammonium complex, benzalkonium chloride (BAC), a general biocide. However, it is not known whether the apparently marginal negative effects on algae growth were directly related to the biocide itself, or indirectly, because of the potential negative effects it would be expected to have on some or all of the phycosphere bacterial community. In general, there was no significant difference between the biomass productivity of the algal grown in the BAC-treated reactors or in the untreated (negative) control reactors (Additional File 6: Fig. S4a). However, this result bears some uncertainty because during the time in the cultivation season during when reactors were treated with BAC, the outdoor temperatures were higher than several weeks earlier when reactors contained the BAC-untreated cultures (negative control) (Additional File 6: Fig. S4b). An additional caveat is that typically higher temperatures result in faster growth and increased susceptibility of C. sorokiniana to attack by V. chlorellavorus. The hypothesis that algal growth rate was indirectly affected by changes in bacterial community is not supported by the ANCOM results (Fig. 10c) that indicated an overall suppression of the most significantly-affected bacteria, albeit, the latter ASVs did not show a strong relationship to biomass productivity. The distribution of the taxonomic assignments of ASVs significantly affected by the BAC treatment was similar to the overall data set, except for the bacteria assigned to the phyla, Firmicutes, for which only a single ASV was significantly changed by BAC treatment, despite being the third most commonly assigned taxa in the collective data set (Table 4). The Firmicute bacteria, Listeria monocytogenes, encodes an efflux pump that confers resistance to BAC (Kovacevic et al., 2016), offering a mechanism by which members the phylum could have feasibly avoided the potentially toxic effects of the BAC treatment.
Table 4
Comparison of bacterial phyla significantly changed by benzalkonium chloride treatment to total amplicon sequence variant taxonomic assignments
| Phylum | Total counts | Total proportion (%) | BAC altered counts | BAC altered proportion (%) |
| Proteobacteria | 462 | 50.55 | 66 | 61.68 |
| Bacteroidetes | 209 | 22.87 | 22 | 20.56 |
| Firmicutes | 59 | 6.46 | 1 | 0.93 |
| Verrucomicrobia | 48 | 5.25 | 7 | 6.54 |
| Armatimonadetes | 7 | 0.77 | 2 | 1.87 |
| Unassigned | 37 | 4.05 | 2 | 1.87 |
| Chlamydiae | 10 | 1.09 | 2 | 1.87 |
| Planctomycetes | 21 | 2.30 | 2 | 1.87 |
| Cyanobacteria | 11 | 1.20 | 2 | 1.87 |
| Actinobacteria | 16 | 1.75 | 0 | 0.00 |
| Patescibacteria | 11 | 1.20 | 1 | 0.93 |
| Gemmatimonadetes | 5 | 0.55 | 0 | 0.00 |
| Acidobacteria | 4 | 0.44 | 0 | 0.00 |
| BRC1 | 3 | 0.33 | 0 | 0.00 |
| Deinococcus-Thermus | 3 | 0.33 | 0 | 0.00 |
| Dependentiae | 3 | 0.33 | 0 | 0.00 |
| Spirochaetes | 2 | 0.22 | 0 | 0.00 |
| Tenericutes | 2 | 0.22 | 0 | 0.00 |
| Chloroflexi | 1 | 0.11 | 0 | 0.00 |
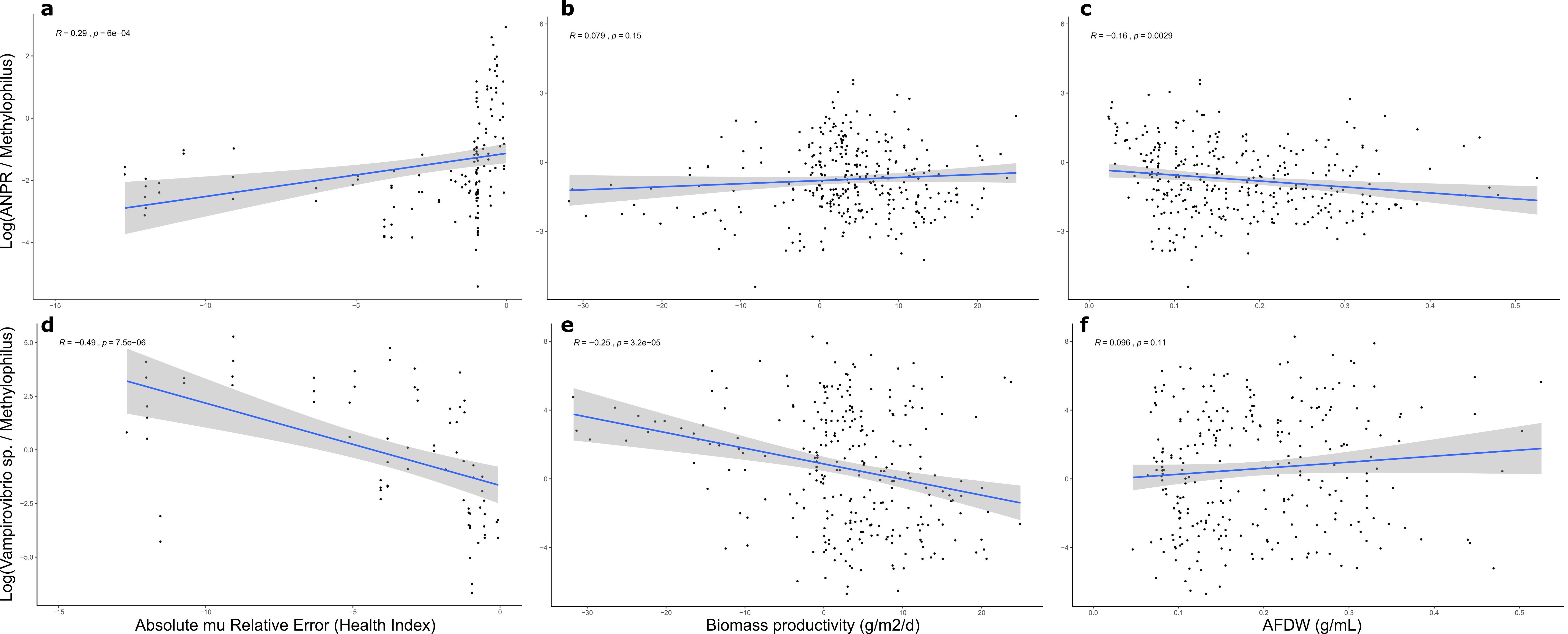
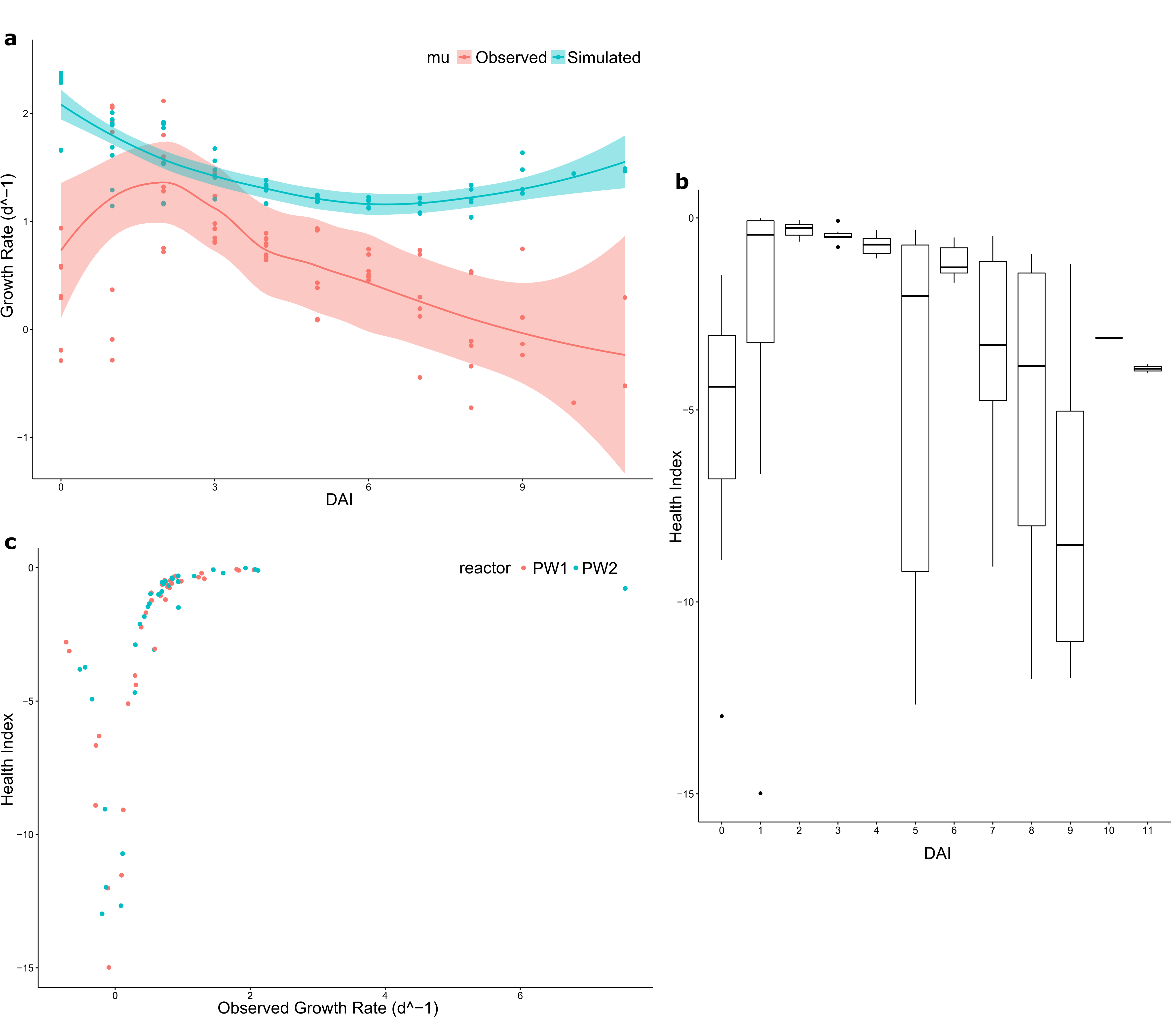
Taking quantitative measurements of algal health in outdoor reactor cultures is challenging. On a regular basis, algal cultures are exposed to continuously changing environmental conditions, particularly light intensity and water temperature. Consequently, the growth of the algae and microorganisms that comprise the phycosphere and their interactions are dynamic. Also, algal growth is affected by its own properties and capacity to adapt to and flourish in the collective environment. For example, a ‘heathy’ dense algal culture would be expected to grow more slowly than a healthy, low-density culture in part owing to increased self-shading. The algal Health Index (HI) computed in this study showed promise for the ability to identify significant bacterial contributors to growth of the target alga. The HI was found to be more sensitive to identifying bacterial correlations in that there were more significant linear correlations between the pathogen V. chlorellavorus and the putative mutualist bacteria, compared to the biomass productivity and AFDW (Additional File 9: Fig. S7). While the latter two metrics are widely used for assessing industrial biomass production goals, applying the HI calculation developed in this study provided a more accurate depiction of biological fitness of the algal populations analyzed here.
The goal of the RAFT project was to develop a global cultivation system for the efficient production of algal biomass. The microbial members of the phycosphere that were identified in this study and associated with algal health, are possible targets for further assessment. The means by which they influence algal health, whether directly or indirectly, requires further study, including specifically how they function and whether they can be utilized in culture augmentation. Further, some of these taxa may prove to be reliable indicators of productivity for C. sorokiniana and for other algal species as well. In the near term, culture-independent analysis of the entire bacterial community is not well-suited for monitoring, and so the design of more specific assays is needed to enable their easy adaptation for field use. For example, a qPCR assay could be developed to monitor Rhizobiales using the 16S rRNA gene sequence identified in this study as having a tight association with algal growth. These approaches can feasibly provide the ability to obtain rapid, quantitative measures of algal health for the incorporation into models, along with other parameters, that will sound a warning or an alert indicating a need for intervention. With the increasingly economical costs of PCR-amplification and DNA sequencing for routine monitoring and microbiome analysis, the phycosphere community composition can be routinely monitored in reactors and the ‘sentinel ASVs’ easily identified by specific qPCR amplification. Further, modelling cultures with microbiome analyses coupled to algal Health Index can facilitate more complete assessment of algal culture health than is presently achieved by measuring algal density alone.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}