VNO sequencing, assembly & V1R recovery
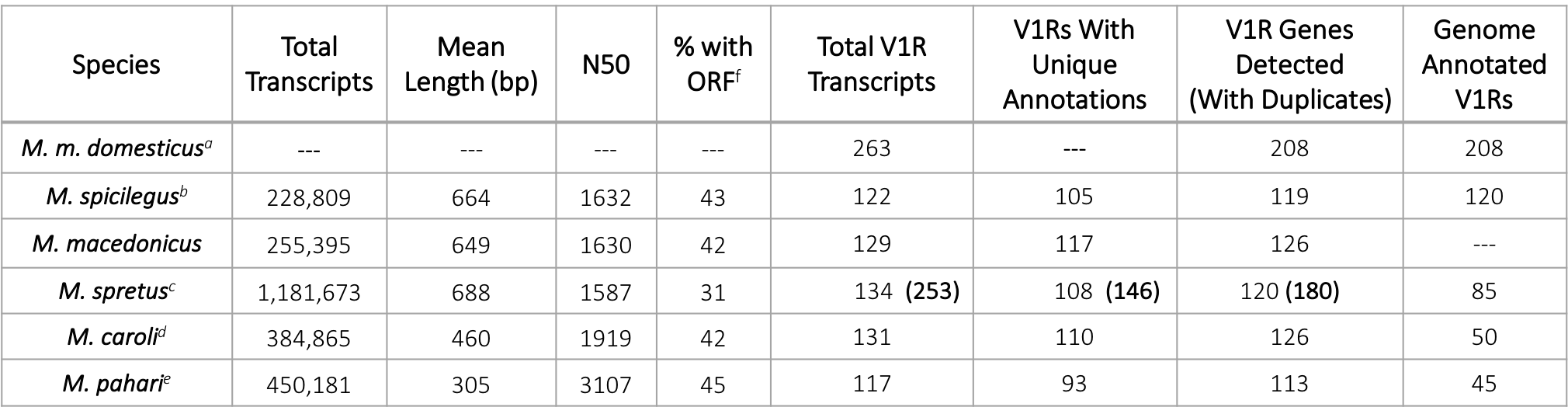
Using wild-derived inbred mouse lines, we characterize V1R repertoires for five Mus species of varying evolutionary distance from the house mouse (1.5-7 mya, Figure 1) by sequencing their VNO transcriptomes using short-read platforms. By sequencing both males and females from inbred mouse lines our aim was to characterize the V1R gene family for each species, rather than differential gene expression or within-species variability, and subsequently compare those data to the house mouse reference genome. The final transcriptome assemblies for each species are of good quality (Table 1). We detect approximately twice the number of V1Rs than are currently annotated in the genomes of M. spretus, M. caroli, and M. pahari and provide the first M. macedonicus V1R dataset (Table 1). The number of V1Rs identified in M. spicilegus is in good agreement with existing genome annotations (Table 1). For one species (M. spretus), the short-read sequencing was performed at greater depth, and an additional round of long-read sequencing was done. This allows us to examine the effectiveness of short versus long-read sequencing for assembling large and highly duplicated gene families such as V1Rs. The total number of assembled transcripts is greater for the M. spretus short-read dataset, as expected from greater sequencing depth (Table 1).
On average, 126 V1R transcripts are recovered from each species’ short-read assembly (Table 1). A subset are transcript variants or gene duplicates, with homology to the same gene in the mouse reference genome (GRCm38.p6). The majority of V1Rs are single-exon genes, however, a substantial number contain introns and express transcript variants (Table 1 & Figure 2) [38]. For a conservative estimate of V1R genes, only unique transcript annotations are included (Table 1). When putative gene duplicates are added, the number of V1R genes increases markedly (Table 1). Compared to the house mouse, the 5 sequenced Mus species have smaller V1R repertoires, consistent with V1R gene expansion in the house mouse (Table 1). However, the addition of long-read sequencing for M. spretus increases the number of V1Rs genes detected, resulting in a repertoire size similar to the house mouse (Table 1). Therefore, whereas the M. spretus V1R repertoire is likely close to complete, long-read sequencing may detect additional V1Rs in M. spicilegus, M. macedonicus, M. caroli and M. pahari. Importantly, our analysis of V1R evolution in Mus is based on (1) a well-annotated mouse reference genome, (2) a comprehensive M. spretus V1R dataset, and (3) >100 V1Rs for all 6 Mus species. Therefore, small gaps in detection across the entire V1R family should not bias the broad patterns of V1R evolution reported here. Furthermore, the discrepancy in repertoire size between the house mouse and other species appears largely accounted for by a putative house mouse specific gene expansion, discussed in further detail below.
V1R evolution across Mus species
To explore V1R evolution, we characterize which receptors share a common ancestor (i.e. are orthologous) by examining relationships within a V1R gene tree containing six Mus species (the 5 sequenced species and the house mouse reference, Additional File 1). A subset of receptors do not exhibit a clear orthologous relationship to any V1R annotated in the mouse reference genome and are classified as non-orthologous genes, indicating either gene loss in the house mouse lineage or lineage-specific expansions in other species (Figure 2). Similarly, a set of receptors annotated in the mouse reference genome was not detected in any other species, suggesting recent expansion in the house mouse lineage (Figure 2).
We classify V1Rs into three broad categories based on their orthologous relationships: (1) V1Rs present only in the mouse reference genome, (2) non-orthologous V1Rs found in species other than the house mouse, and (3) V1Rs with orthology across multiple species. V1Rs with orthology across multiple species are further categorized based on the number of species represented in each orthologous receptor group (orthogroup). Orthogroups with 2-3 species are classified as “low orthology,” and orthogroups with 4-6 species as “high orthology” (Figure 2A). The majority of transcripts have some evidence for orthology (88.5%, Figure 2A). Furthermore, most transcripts are highly orthologous (75.1%, Figure 2A), indicating that missing V1Rs are unlikely to bias broad patterns identified here. Although many receptors are shared across species, approximately 25% of all V1R transcripts, and 59% of all unique V1R annotations, are either low orthology, non-orthologous, or present only in the mouse reference genome (Figure 2A). This indicates that the dramatic V1R gene turnover observed among more divergent mammalian species, such as across tetrapods or between rodent species [19, 21, 23], is replicated within the genus Mus, albeit on a more limited scale. Furthermore, we find a little over 5% of total V1Rs are present in only the house mouse reference genome. Interestingly, nearly all of these reference-only receptors are located in a single clade and are tandemly arrayed on a single chromosome, suggesting a potential house mouse specific expansion.
We next examine the presence of gene duplicates and transcriptional variation across species (Additional File 2). A similar proportion of V1R gene duplicates are identified across all 5 species (10-16%, Figure 2B). The proportion of V1R transcript variants detected is also comparable across species, with the clear exception of M. spretus (Figure 2B). As expected, the addition of long-read (M. spretus) sequencing data recovers many more transcript variants than short-read sequencing datasets (Figure 2B). Interestingly, the same number of V1R genes expressing distinct coding transcript variants are detected in M. spretus as in the house mouse (43 V1R genes, Additional File 3: Figure S1). However, the identity of V1Rs exhibiting alternative spliceforms, and the clades they belong to, vary between the two species (Additional File 3: Figure S1). In contrast, the proportion of gene duplicates detected is similar between M. spretus and the other species. This indicates that, for gene families such as V1Rs, short-read datasets are sufficient for identifying gene duplicates.
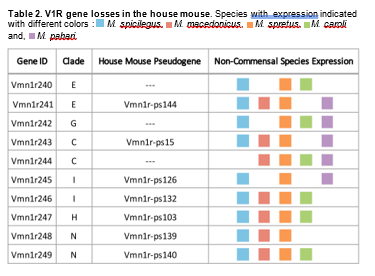
Our characterization of V1R repertoires across Mus species allows for a reliable estimate of V1R gene loss in the house mouse. We detect evidence for 10 such gene losses, distributed across six clades (Table 2 & Figure 3A: indicated in red text). All V1R genes lost in the house mouse are present in at least 3 of the 5 sequenced Mus species, including close relatives (Table 2). Most gene losses have corresponding pseudogenes in the house mouse (Table 2). It appears gene losses are relatively uncommon compared to the abundant gene gains, at least within the house mouse lineage.
Novel V1R clade: clade “N”
In addition to the house mouse gene losses observed in clades E, C, H, I and G, we identify a novel V1R clade (Table 2, Figure 3A). This novel clade “N” has been lost in the house mouse and consists of two receptor orthogroups. Both clade N receptors (Vmn1r248 and Vmn1r249) are expressed in at least three Mus species (Additional File 3: Figure S2) and have corresponding pseudogenes in both the house mouse (M. m. domesticus) and the rat (Rattus norvegicus).
Variable patterns of evolution across V1R clades
Gene Turnover: Orthology, Duplication & Repertoire Size
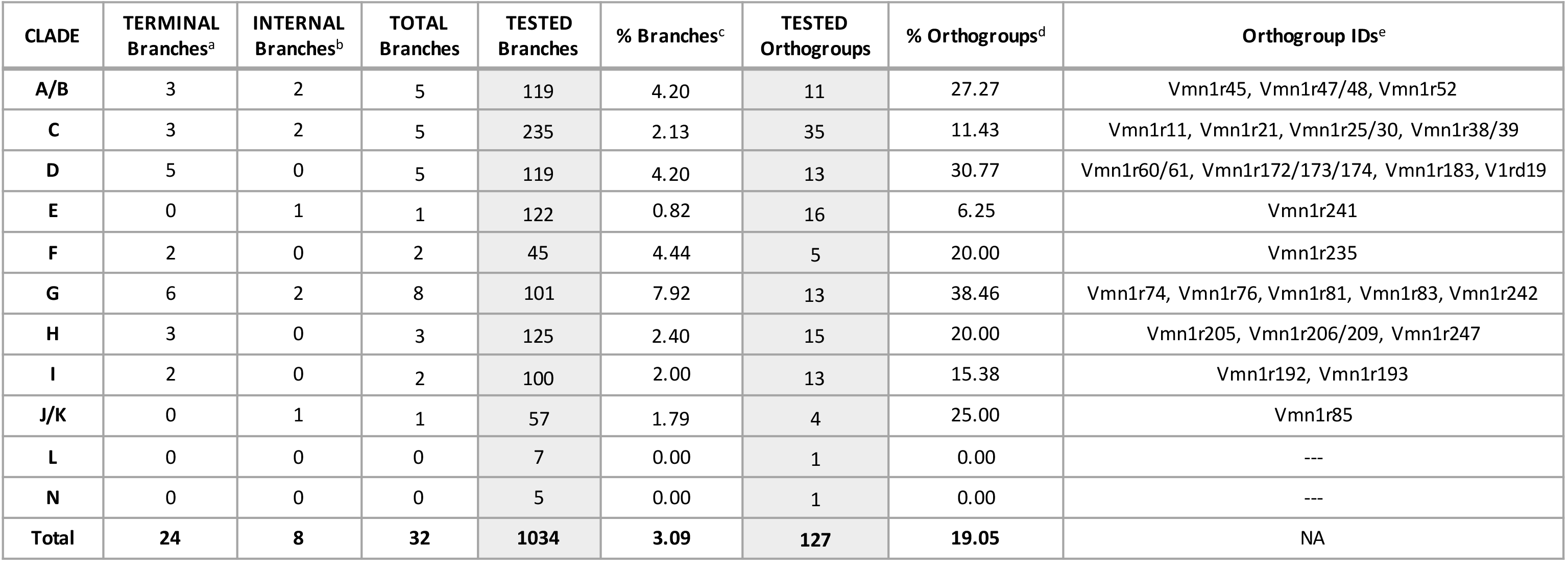
The maintenance or loss of gene orthologs is a major mode of chemosensory evolution [19, 21, 23, 27]. High degrees of gene orthology across species suggests distinct evolutionary trajectories compared to clades exhibiting lineage-specific gene expansions. Patterns of V1R gene orthology and duplication vary across clades. Four of the 11 clades are very orthologous (E, F, J/K and L: >80% of receptors are high-orthology), with clade G trailing behind with more non-orthologous receptors (Figure 3A, B). Each of these clades has 5 or fewer gene duplicates detected, however, the proportion of duplicates is variable (Figure 3C, D). Clades E, F and G have very low proportions of gene duplicates, while clade J/K has among the highest (Figure 3C).
Clades C, D and H have abundant low-orthology and non-orthologous receptors (Figure 3B), indicating greater evolutionary lability. While most orthologous relationships are straightforward, some orthogroups contain multiple house mouse receptors, and are annotated with combination-IDs to indicate the relationship to multiple genes (e.g. Vmn1r25/30). These receptor groups are the result of one or more duplication events within the Mus lineage, and are unequally distributed across clades, with 76% located in clades C and D (Figure 3A). In addition, all reference-only V1Rs are located in these same two clades (Figure 3B). Not surprisingly, clades C, D and H have the highest number of detected gene duplicates (19 or more) and have similarly high proportions of duplicates by clade size (Figure 3C, D). Thus, all three clades have evidence for substantial gene expansions, particularly clade D within the house mouse lineage.
We examine V1R clade sizes across all 6 species. With the striking exception of clade D, the house mouse clade sizes are very similar to the 5 other species, (Figure 4). This general pattern provides further evidence that receptor recovery is high and species’ repertoires are near complete. Interestingly, the M. spretus repertoire is largest for several clades (A/B, C, E, H and I; Figure 4), indicative of M. spretus-specific gene expansions.
Fast-evolving clades
Clade H
Clade H appears to be a mouse-specific V1R expansion, as it’s absent in the rat genome [21]. The clade is characterized by patterns of low orthology, abundant gene duplicates, and variable repertoire size across species (Figures 3 & 4). In contrast to this pattern of high gene turnover, relatively few clade H branches have evidence for positive selection (Table 3). A sub-region of clade H containing Vmn1r217, 219 and 220 receptors exemplifies the pattern of low orthology, while the receptor orthogroup Vmn1r206/209 is representative of the abundant gene duplicates (Figure 5A). Intriguingly, the Vmn1r206/209 orthogroup also has evidence for positive selection, pointing to particularly strong diversifying selection within this receptor group (Table 3). A striking exception to the patterns of dynamism observed in clade H is the extremely conserved receptor group Vmn1r197 (Figure 5A). The general pattern of rapid species-specific gene gains and losses suggests clade H receptors may play an important role in detecting complex species-specific signals.
Clade C
Clade C is the largest V1R clade across species with the exception of the house mouse. Clade C also exhibits variable repertoire sizes across species, suggestive of lineage-specific evolution (Figure 4). This inference is supported by the large numbers of combination-ID orthogroups, gene duplicates, non-orthologous receptors, and house mouse-specific gene gains (Figure 3). In contrast to the high levels of gene turnover, relatively few clade C branches have evidence of positive selection (Table 3). The phylogenetic structure of clade C comprises three sub-clades, one of which is quite orthologous (Figure 5B). Interestingly, the non-orthologous receptors are largely clustered in one sub-clade (57%, Figure 5B), while the majority of receptors under positive selection are located another (21% orthology, Figure 5B). Together, this suggests these subclades may be experiencing distinct forms and rates of receptor evolution. Two clade C receptors, Vmn1r9 and Vmn1r10, have been implicated in pup odor detection in house mice [42]. However, these receptors also respond to female odors, and thus may detect chemosensory components of the nest environment [42]. These two receptors are part of a single receptor orthogroup (Vmn1r9/10) that is both orthologous and highly duplicated (Figure 5B). The sister group Vmn1r7/8 exhibits a similar pattern of high orthology and abundant duplication (Figure 5B). Given the potential role of Vmn1r9/10 receptors in pup odor detection, and the lineage-specific evolutionary patterns observed in Vmn1r7/8 and Vmn1r9/10, these receptor groups are interesting candidates for future functional tests of their role in conspecific chemosignaling.
Clade D
Clade D exhibits a large skew in repertoire size within the house mouse (Figure 4), and has the most dramatic patterns of non-orthology of all V1R clades (Figure 3B). Nearly all reference-only V1Rs (50/53: 94%) are located in clade D, providing further support for a large recent gene expansion in the house mouse, despite the potentially low receptor recovery of other species in this clade (Figure 3B). These receptors are similar in sequence and cluster together on chromosome 7, consistent with recent tandem gene duplication. While we do not find evidence for a comparably large expansion in the other mouse species, we recover approximately twice as many clade D receptors in M. spretus relative to the other four species (Figure 4). It is possible that similar expansions exist in the other species that are not detected here, particularly given prior evidence that clade D receptors are very lowly expressed in the house mouse [38]. Additionally, clade D has a high proportion of non-orthologous receptors and gene duplicates. In line with the pattern of high gene turnover, clade D also has a large percentage of orthogroups under positive selection (30.77% of tested orthogroups, Table 3). Given the evolutionary labile nature of clade D, there are a few receptors that stand out as highly orthologous (V1rd19, Vmn1r179 and Vmn1r172/173/174, Figure 5C), two of which have evidence for positive selection (Table 3). Clade D thus appears to be experiencing lineage-specific evolution at the scale of both gene gains and losses and as well as sequence divergence.
Conserved clades & female conspecific detection: Clades E & F
A subset of V1R clades are highly conserved, and thus good targets for uncovering receptors with conserved olfactory functions. Clades E and F are characterized by high orthology (Figure 3B), long internal branch lengths and short terminal branch lengths, suggestive of old gene duplications maintained within the Mus lineage (Additional File 3: Figure S3). In contrast, very few recent gene duplications are detected (Figure 3C, D). In addition, clade E has the lowest proportion of branches under positive selection of any clade (Table 3). Clade F, on the other hand, has a single receptor (Vmn1r235) with evidence for positive selection in two species (Table 3, Additional File 3: Table S1). A subset of 5 clade E receptors is important for the detection of female-specific urine odors in house mice (Vmn1r68, Vmn1r69, Vmn1r71, Vmn1r184, Vmn1r185) [40]. Two clade E sub-regions containing these same 5 receptor groups are shown in Figure 5D, E; those with the strongest support for female odor detection are highlighted in red (Vmn1r69 and Vmn1r185) 40]. Vmn1r68 and Vmn1r69 are sister to each other in the gene tree and are highly orthologous, however, Vmn1r69 has no orthologs detected among the more basal species (M. caroli and M. pahari; Figure 5D). It is plausible that Vmn1r69 is the result of a gene duplication event preceding the divergence of the four more derived species (Figure 5D), providing enhanced specificity or sensitivity toward female-specific urine odors. The second clade E sub-region contains receptors: Vmn1r184, Vmn1r185, and Vmn1r71. Vmn1r184 and Vmn1r185 are sister receptor groups, in which Vmn1r185 is highly orthologous and Vmn1r184 appears to be the result of a recent duplication event (Figure 5E). Interestingly, Vmn1r184 is detected in only the house mouse and M. spicilegus (Figure 5E). Furthermore, M. spicilegus has evidence for a species-specific Vmn1r184 duplicate, and has an absence of Vmn1r185 expression (Figure 5E). The distinct expression pattern of Vmn1r184 in M. spicilegus is noteworthy given this species’ unique social structure, which includes cooperative behaviors and social monogamy [57]. In comparison, Vmn1r71 is highly orthologous (Figure 5E), but displays remarkable transcriptional variability, most of which is located at either the C-terminus or N-terminus regions of the protein (Additional File 3: Figure S3). Broadly, clades E and F display patterns of conservation, with some evidence of positive selection in clade F, and potential lineage-specific gains in clade E among receptors involved in detecting female cues.
Clade J/K evolution & the detection of estrus cues
Clade J/K is a small clade of only 4 receptor groups that is both highly orthologous and boasts one of the highest proportions of gene duplicates (Figure 3). This clade thus encompasses a unique mixture of conservation and divergence, in which there is very little gene loss but gene gains are abundant (Figures 3 & 5F). Clade J/K is also the only clade for which half of the receptors have known ligands [40, 49]. In the house mouse, two of the four J/K receptors have been shown to detect estrus cues (i.e. sulfated estrogens) in female urine [40, 49]. Given the unique features of this clade, we examined in greater detail the amino acid changes across species within the two deorphanized receptors (Vmn1r85 and Vmn1r89). The Vmn1r89 receptor group has evidence for short and long transcript types across Mus species (Additional File 3: Figure S4). Many species have only one form detected. However, the house mouse and M. spretus express both forms as transcript variants, while M. pahari appears to have distinct genes generating these two forms (Additional File 3: Figure S4). The widespread detection of both transcript types suggests they may facilitate the detection of distinct ligand (i.e. sulfated estrogen) features. This is particularly compelling given that in the house mouse, Vmn1r89-expressing VSNs detect multiple sulfated estrogen molecules and are more broadly tuned than Vmn1r85-expressing VSNs [40]. In comparison, the Vmn1r85 receptor group is highly conserved among the 3 Mus species most closely related to the house mouse (Figure 5F), with the majority of substitutions concentrated in M. caroli and M. pahari (Additional File 3: Figure S5). For both Vmn1r85 and Vmn1r89, the highest proportion of amino acid site changes detected across species occurs in extracellular regions (Additional File 3: Figure S6). The trend towards greater extracellular substitutions is consistent with a prior analysis of molecular evolution in 22 V1Rs, demonstrating that most sites with evidence for positive selection are located in extracellular motifs [30]. Moreover, positive selection is detected in Vmn1r85 (Table 3) at an internal branch containing the house mouse and close relatives (M. spicilegus, M. macedonicus and M. spretus, Additional File 3: Table S1).
{kind=link}
{kind=link}
{kind=link}