3.1. Functional enrichment analysis of differentially expressed necroptosis-related genes
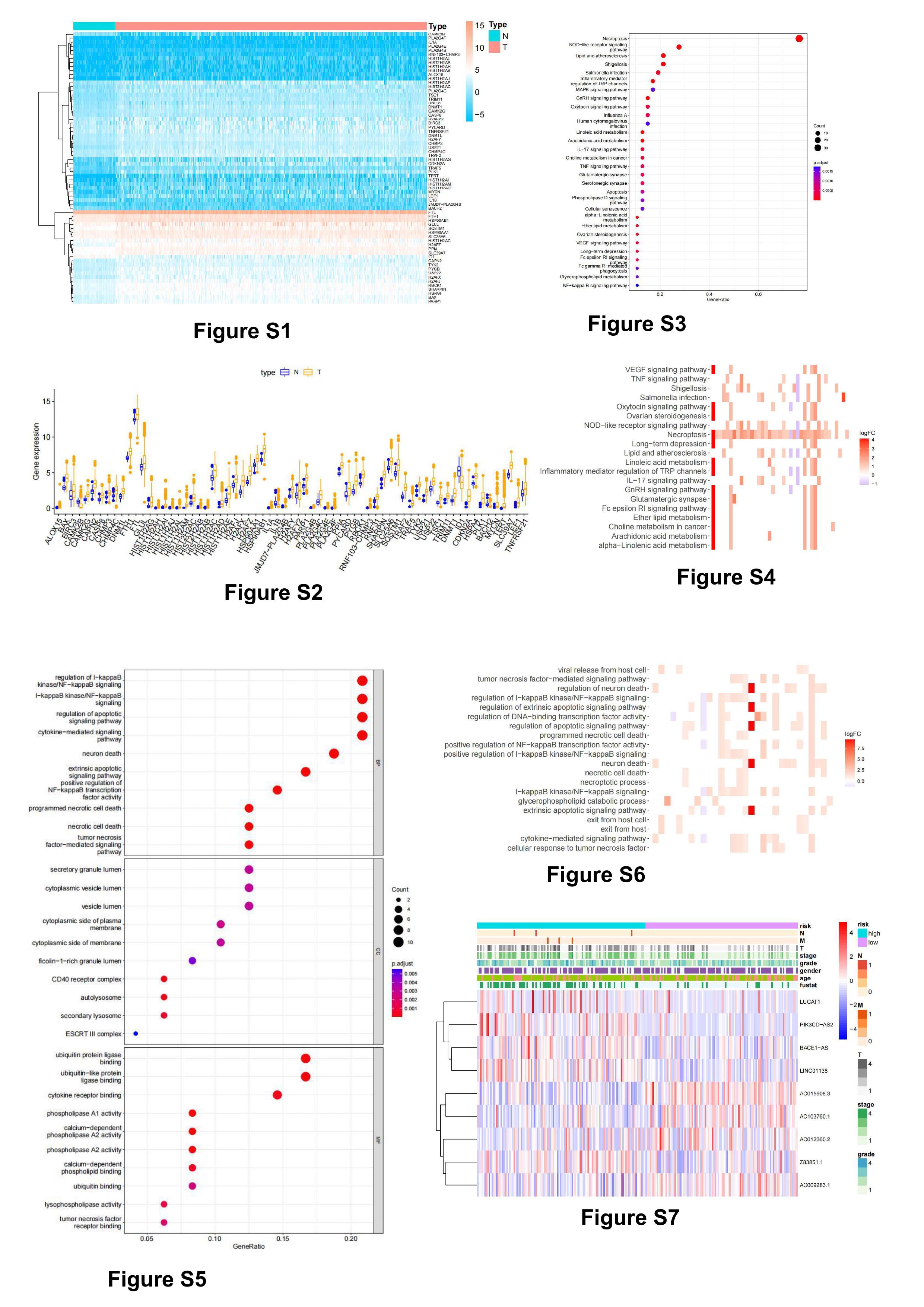
67 necroptosis-related DEGs were obtained by differential analysis of the data screened from the TCGA database, including 63 up-regulated genes and 4 down-regulated genes (figure 2A, S1, S2). We further analyzed the regulatory relationship between these differentially expressed genes(figure 2B). These genes were then subjected to KEGG pathway and GO enrichment analysis. The KEGG enrichment analysis showed that the DEGs were mainly enriched in Necroptosis, NOD-like receptor signaling pathway, Linoleic acid metabolism, Inflammatory mediator regulation of TRP channels, alpha-Linolenic acid metabolism, Lipid and atherosclerosis, GnRH signaling pathway, Shigellosis, Arachidonic acid metabolism (figure 2C, S3, S4). GO enrichment analysis showed that In the biological process category, the DEGs were mainly enriched in regulation of I-kappaB kinase/NF-kappaB signaling, I-kappaB kinase/NF-kappaB signaling, programmed necrotic cell death, In the cellular components category, the DEGs are mainly enriched in CD40 receptor complex, autolysosome, secondary lysosome, In the molecular function category, the DEGs are mainly enriched in phospholipase A1 activity, calcium-dependent phospholipase A2 activity, ubiquitin protein ligase binding(figure 2D, S5, S6).
3.2. The necroptosis-related lncRNA predictive signature
The lncRNAs were isolated from the DEGs, and finally 765 necroptosis-related lncRNAs (Table S1) were obtained, and then univariate COX regression analysis was used to obtain 94 lncRNAs related to prognosis, and multivariate COX analysis was used to obtain the final 9 lncRNAs. The expression of these 9 necroptosis-related lncRNAs were then visualized(figure 2E), Then using Cytoscape and the ggalluvial R software package to find co-expression mRNAs for further visualization(figure2F,|R2|>0.4and p<0.001). 31 genes co-expressed with these 9 lncRNAs. Among them, BACE1-AS, LINC01188, LUCAT1, PI3KCD-AS2 and Z83851.1 were risk factors, AC009283.1,AC012360.2, AC015908.3 and AC103760.1 were protective factors (figure 2G). Then we analyzed the expression of these co-expressed genes in cancer tissues and adjacent tissues. The results indicated that BAX,CDKN2A,FTH1,H2AF,FTL,HSP90AB1, HSPA4, PLK1 and TRAF2 were highly expressed in tumor tissues, 6 of these genes were significantly associated with the prognosis of hepatocellular carcinoma(figure 2H-J, Immunohistochemical results from proteinatlas database(https://www.proteinatlas.org/). gene expression profile from GEPIA database(http://gepia.cancer-pku.cn/index.html).). The formula for calculating the risk score is: Risk score=(0.597×BACE1-AS expression)+(-0.537×AC015908.3 expression)+(0.342×PIK2HD-AS2 expression)+(-0.491×AC012360.2 expression)+(0.520×Z83851.1 expression)+(-0.664×LINC01138 expression)+(-0.457×AC103760.1 expression)+(-1.112×AC009283.1 expression) +(0.340×LUCAT1 expression).
3.3. Validation of the necroptosis-related lncRNA predictive signature
Calculateing the risk score of all patients, Then we divided patients into two groups by the median risk score. As risk scores increased, so did the number of patient deaths (figure 3A). We then performed PCA principal component analysis, which showed that patients grouped according to risk lncRNA was more specific than all gene, all lncRNA, and necroptosis-gene (figure 3B). Kaplan-Meier analysis method was used to analyze the OS time of high-risk group and low-risk group to determine the value of risk score in predicting the prognosis of patients with hepatocellular carcinoma. The result showed that the OS of the high-risk group was significantly shortened (figure 3C, p<0.001). Subsequently, Univariate COX regression analysis showed that tumor stage, T grade, N grade and risk score were the factors affecting the prognosis of patients (figure 3D, p<0.05). Multivariate COX regression analysis showed that only risk score was an independent risk factor affecting the prognosis of patients (figure 3E, p<0.05). According to the drawn ROC curve, it was found that the AUC of the risk score was 0.874, indicating that the risk score is better than other clinical features for the prognosis of hepatocellular carcinoma patients (figure 3F). And the AUC of 1-year, 3-year and 5-year were 0.8, 0.759, 0.787, indicating that the risk score has good accuracy in predicting survival time(figure 3G). Differences in lncRNA expression between high- and low-risk groups were analyzed according to clinical characteristics (figure S7). A nomogram was constructed by clinical features and risk score, which predicts 1-, 3-, and 5-year prognosis in patients with hepatocellular carcinoma (figure 3H). Calibration curves show good agreement between predicted 1-, 3-,and 5-year survival rates and patients' actual OS (figure 3I).
3.4. The accuracy of the predictive signature in predicting the prognosis of hepatocellular carcinoma patients with different clinical characteristics
In order to verify the accuracy of the Predictive Signature in predicting the prognosis of hepatocellular carcinoma patients with different clinical characteristics, hepatocellular carcinoma patients were grouped according to age, gender, stage, T stage and grade. The OS of low-risk group were significantly higher than the high-risk group(figure 4A). These results suggest that predictive features can predict the prognosis of patients with hepatocellular carcinoma regardless of clinical features.
3.5. Validation of predictive signature
To verify the validity of the predictive signature, the TCGA data were randomly divided into two cohorts, and the clinical characteristics of two cohorts were shown in Table 1. The results were the same as expected, and the OS of patients in the high-risk group in the first cohort was significantly lower than that in the low-risk group (figure 4B,p<0.01). Another cohort also showed the same result(figure 4C,p<0.01). And the AUC of the ROC curve for the first cohort at 1-, 3-, 5- year were 0.851, 0.804, 0.802 (figure 4D). The AUC of the ROC curve for another cohort at 1-, 3-, 5- year were 0.735, 0.716, 0.765 (figure 4E). Box plots were then drawn according to different clinical characteristics and risk scores(figure 5A), This suggests that there are differences in risk scores for different clinical features.
Table 1
|
Variables
|
Entire
TCGA dataset(n =342)
|
Validation cohort
|
|
First cohort(n=172) second cohort(n=170)
|
|
Age(%)
<=65
>65
|
216(63.16)
126(36.84)
|
109(63.37)
63(36.63)
|
107(62.94)
63(37.06)
|
|
Gender(%)
Female
male
|
109(31.87)
233(68.13)
|
55(31.98)
117(68.02)
|
54(31.76)
116(68.24)
|
|
Grade
G1
G2
G3+4
Unknow
|
53(15.5)
161(47.08)
123(35.96)
5(1.46)
|
25(14.53)
87(50.58)
58(33.72)
2(1.16)
|
28(16.47)
74(43.53)
65(38.24)
3(1.76)
|
|
Stage(%)
I+II
III+IV
Unknow
|
238(69.59)
83(24.27)
21(6.14)
|
115(66.86)
45(26.16)
12(6.98)
|
123(72.35)
38(22.35)
9(5.29)
|
|
T(%)
T1+2
T3+4
TX+ Unknow
|
252(73.68)
87(25.44)
3(0.88)
|
121(70.35)
50(29.65)
1(0.58)
|
131(77.06)
37(21.76)
2(1.18)
|
|
M(%)
M0+1
MX+ Unknow
|
247(72.22)
95(27.78)
|
121(70.35)
51(26.95)
|
126(74.12)
44(25.88)
|
|
N(%)
N0
NX+ Unknow
|
242(70.76)
100(29.24)
|
120(69.77)
52(30.23)
|
122(71.76)
48(28.24)
|
3.6. Gene enrichment analysis
Differences in prognosis of patients in high- and low-risk groups were investigated using GSEA. We found that the high-risk groups had cell cycle passway, complement and coagulation cascades passway, drug metabolism cytochrome p450 passway, metabolism of xenobiotics by cytochrome p450 passway, oocyte meiosis passway, peroxisome passway, fatty acid beta oxidation, azurophil granule, aromatase activity and purine metabolism passway significant enrichment (figure 5B, 5C), It shows that high-risk patients are closely related to tumors and a variety of substances metabolism-related pathways.
3.7.The correlation between risk scores and immune cells and functions
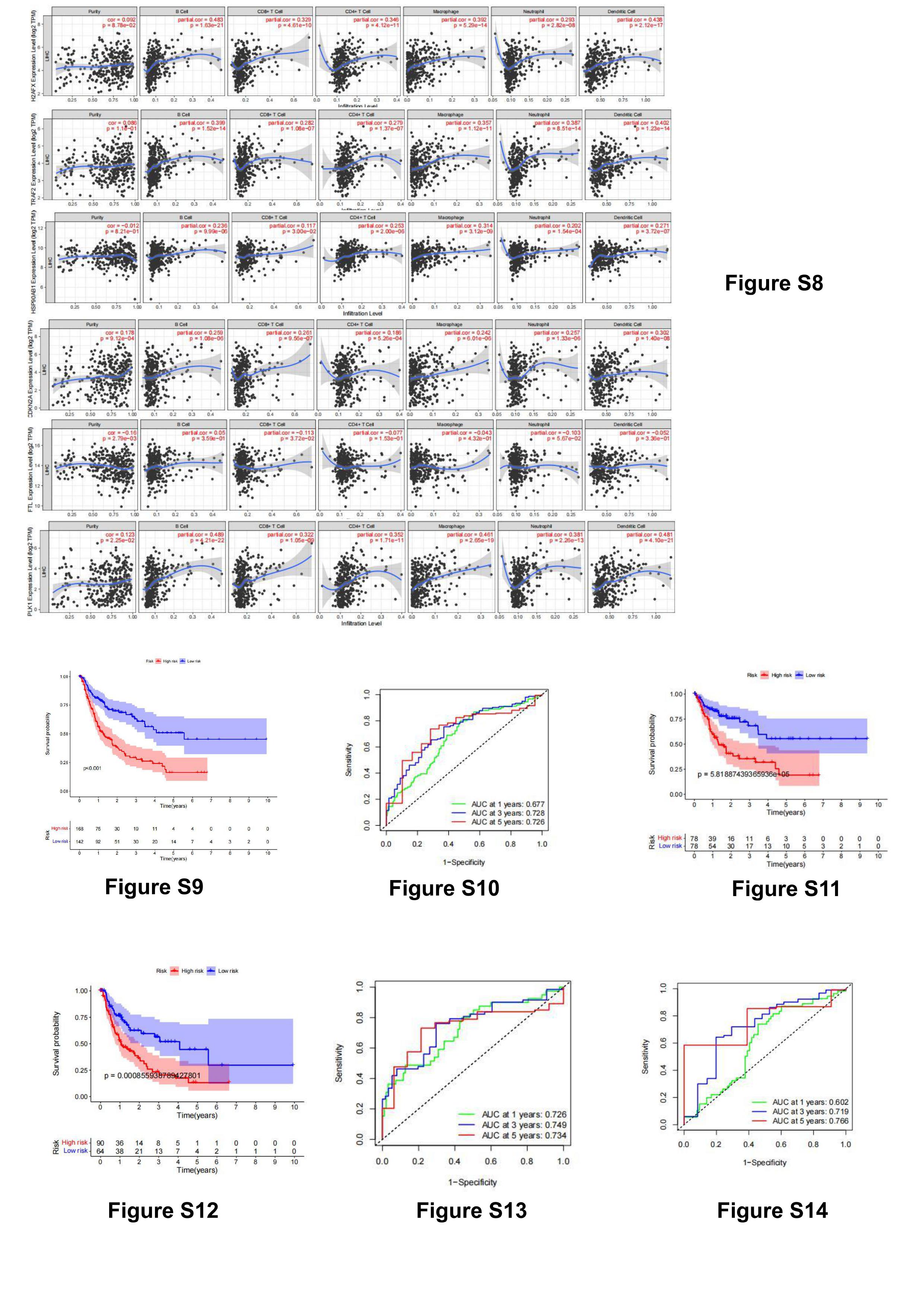
To further explore the correlation between risk scores and immune cells and function, we quantified immune cell subsets and associated functions. The results showed that active dendritic cells (aDCs), macrophages, mast cells, natural killer cells (NK), T regulatory cells (Tregs) were different between high-risk and low-risk groups. salience (figure 6A). Likewise, there were significant differences in immune function between the two groups (figure 6B). Then we analyzed the differences of different immune checkpoints between the two groups, and found that the expressions of CD86, LAIR1, CTLA4, VTCN1,TNFRSF18,CD80,CD276,HHLA2, TNFSF4, TNFRSF8, TNFRSF4, TNFRSF9,LGALS9,HAVCR2 and TNFSF15 were significantly different(figure 6C). To further explore the relationship between 9 necroptosis-related lncRNAs (BACE1-AS, LINC01188, LUCAT1, PI3KCD-AS2, Z83851.1, AC009283.1, AC012360.2, AC015908.3, AC103760.1) and immune cell infiltration, we analyzed their co-expression Expression of necroptosis-related genes in tumor tissue, among which H2AFX, TRAF2,HSP90AB1, CDKN2A, PLK1, and FTL are highly expressed in tumor tissue. The relationship between them and immune cell infiltration was also analyzed online using the TIMER database, and the results showed that they were closely related to a variety of immune cell infiltration(figure S8, immune infiltration level from the TIMER database(https://cistrome.shinyapps.io/timer/).).
3.8. Analysis of Gene Mutation in Patients with Hepatocellular Carcinoma
In order to further explore the reasons for the occurrence and development of hepatocellular carcinoma, we conducted gene mutation analysis(figure 6D). First, we analyzed the genes with the highest mutation rates. The results show that these mutated genes were significantly associated with necroptosis-related genes(figure 6E). We also analyzed the bases of gene mutation and found that the most common T base changed to A base(figure 6F). These mutated genes are enriched in RTK-RAS, WNT, NOTCH, PI3K and other pathways(figure 6G). Subsequent drug-mutated gene correlation analysis showed that the therapeutic effect of DRUGGABLE GENOME and CLINICALLY ACTIONABLE may be better(figure 6H). Finally, we found that the heterogeneity of hepatocellular carcinoma is more obvious, which suggests that the drug resistance of liver cancer will be very strong(figure 6I).
3.9. the necroptosis-related lncRNA predictive signature for DFS
Then we constructed the necroptosis-related lncRNA predictive signature for DFS.We collected DFS data on hepatocellular carcinoma patients from the cBioPortal database, including 310 patients. After univariate Cox regression analysis, 33 necroptosis-related lncRNAs were found to be significantly associated with DFS in patients with hepatocellular carcinoma. After multivariate Cox regression analysis, 7 necroptosis-related lncRNAs were obtained, which were used to construct predictive signatures. Risk score=(0.357×ZFPM2-AS1 expression)+ (-0.332×AC026401.3 expression)+ (0.596×SREBF2-AS1 expression)+ (0.293×AL391427.1 expression)+ (0.405×MIR4458HG expression)+ (0.523×AC005229.4 expression)+ (0.512×LYRM4-AS1 expression). The risk score for each patient was calculated according to the formula, and the patients in the entire dataset were divided into high-risk and low-risk groups based on the median. Kaplan-Meier survival curve analysis showed that the DFS in the high-risk group was significantly shorter than that in the low-risk group (figure S9,p<0.001). The AUCs for 1-, 3-, and 5-year survival were 0.677, 0.728, and 0.726 (figure S10). To validate the predictive signature, 274 patients were randomized into two cohorts. The validation results were the same as expected, and the DFS of patients in the high-risk group in the first cohort was significantly lower than that in the low-risk group (figure S11,p<0.01). Another cohort also showed the same result (figure S12,p<0.01). And the AUC of the ROC curve of the first cohort at 1-, 3-, 5- year were 0.726, 0.749, 0.734 (figure S13). The AUC of the ROC curve of another cohort could reach 0.602, 0.719, 0.766 at 1-, 3-, 5-year (figure S14).

{kind=link}
{kind=link}