Animals and treatment
C57/BL6 mouse breeding conditions and treatment methods in this experiment strictly complied with the Animal Care and Ethical Committee of Qingdao Agricultural University. The mice were bred in a house with a temperature of 23 ℃, 12 h light/dark cycles (lights off at 19:00 h), and adequate water and feed. Mating was arranged at about 17:00 h, and the presence of a vaginal plug in females the next morning was viewed as a successful mating, which was considered as E0.5.
In the present study, ZEA (Sigma, Z2125, MO, USA) was diluted to 40 mg/ml using DMSO, stored at -20 ℃, and diluted to 40 µg/ml with PBS before use. The dose of ZEA was chosen based on the study by Liu et al [30]. At E16.5, the mother mice were exposed to ZEA orally at a dose of 40 µg/kg body weight (BW) until PD0 or PD3 and were termed the ZEA group. Mother mice that were not treated were called the CTRL group. Subsequently, in order to conduct ZEA toxicity rescue, at E17.0, a part of ZEA group was orally taken CoQ10 (Sigma, C9538) at a dose of 20 mg/kg BW, which was dissolved in corn oil and called ZEA+ CoQ10 group. To eliminate the influence of the corn oil on the experimental results, one part of the control group was orally taken corn oil (the solvent of CoQ10) according to body weight and was designated as the corn oil (CO) group. At the same time, corn oil was also administered to one part of the ZEA group, which was designated as the ZEA+CO group. The dose of CoQ10 was chosen based on a previous study [40].
Sample collection
Sample preparation for sequencing: The method was the same as previously described [33]. Sample preparation for the experiment: At PD0 and PD3, ovaries were separated from mouse ovarian capsules in 0.9% normal saline. For the two ovaries from the same mouse, one was collected and cryopreserved at -80 ℃, and the other one was fixed overnight at 4 ℃ in 4% paraformaldehyde (PFA, Solarbio, P1110, Beijing, China).
scRNA-seq libraries and sequencing
We used the single cell system of the 10× Genomics platform to obtain gel bead emulsions (GEMs). The 10× Genomics Chromium Single Cell 3’ library and Gel Bead Kit v3 was applied for reverse transcription, barcode, and library establishment, according to the manufacturer’s protocol. Paired-end sequencing was performed using an Illumina HiSeq X Ten instrument from Beijing Novogene Bioinformatics Co., Ltd., China. Raw data were then analyzed by CellRanger v.3.1.0 software, and the gene expression matrixes of four ovarian samples were obtained.
Analysis of scRNA-seq data
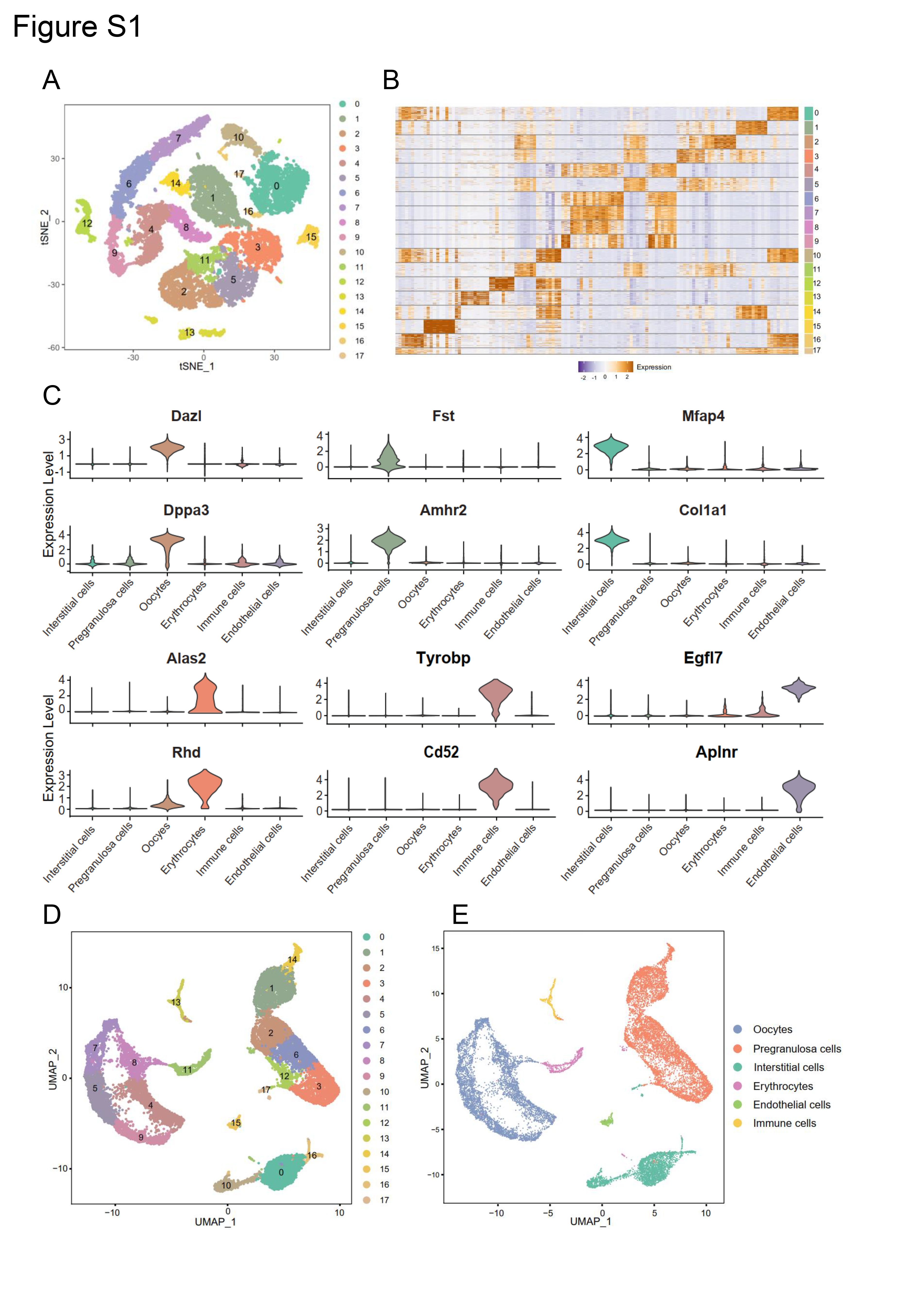
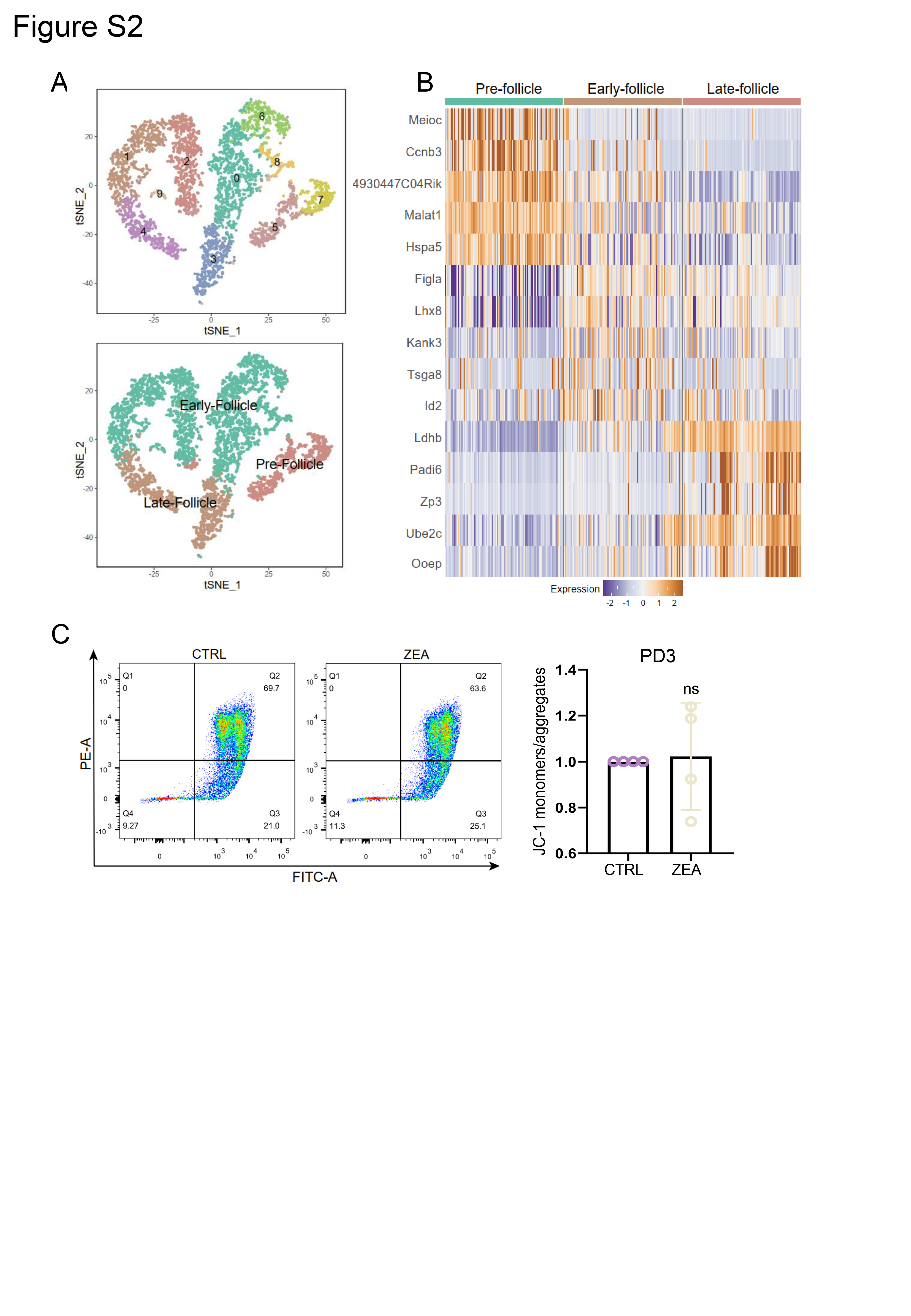
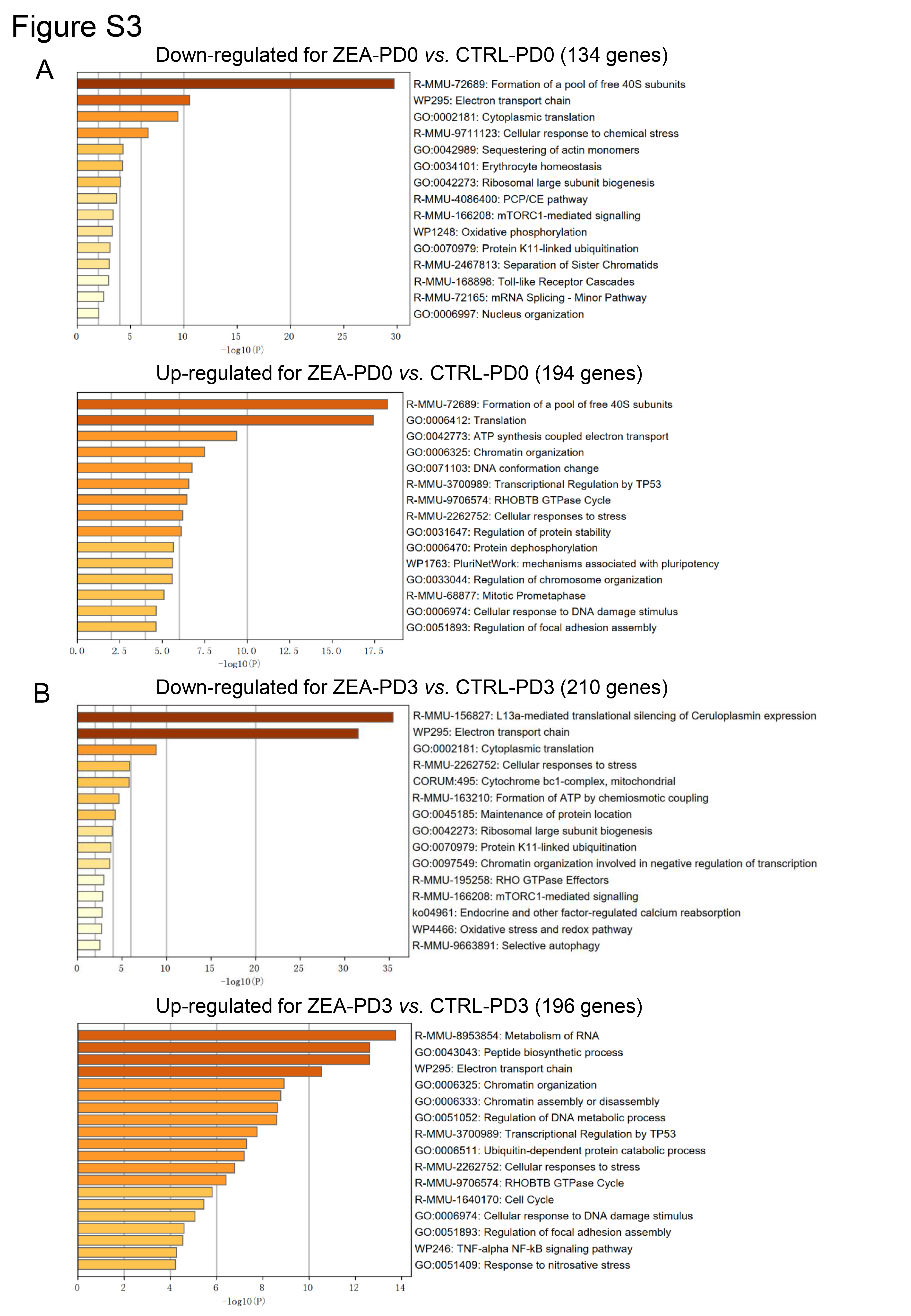
The “DoubleFinder” package was used to filter out abnormal cells (McGinnis et al. 2019), then “Seurat” was used for normalization, integration, and dimensionality reduction of the four sample sets. By using the “RunTSNE” function, the cells were divided into several clusters, and the cell types were determined according to the classic cell markers. The target cell type (oocyte) was extracted for further mining and analysis. The “FindMarkers” function was used to find the differential genes between the ZEA and CTRL groups. Further, metascape (https://metascape.org/gp/index.html) and GO packages were performed for enrichment analysis.
Newborn ovary culture in vitro and RNA interference (RNAi)
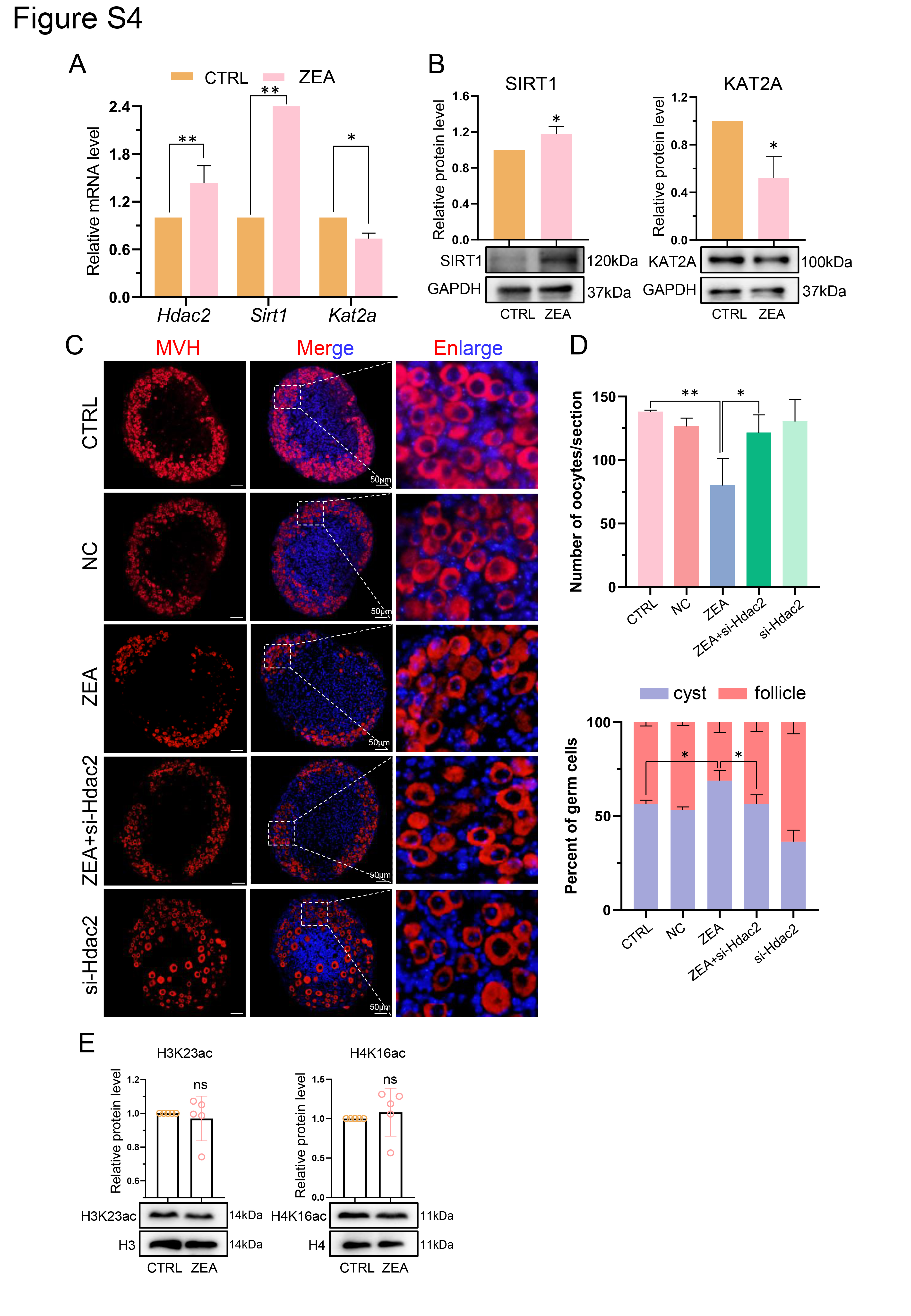
Ovaries isolated from PD0 mice were cultured for 3 days as previously described [9]. The isolated ovaries were cultured in 24-well culture dishes (NEST Biotechnology, Wuxi, China) in ovarian medium, which contained DMEM/F12 (Hyclone, SH30023.01B, Beijing, China), α-MEM (Hyclone, SH30265.01B) (1:1), 10% fetal bovine serum (FBS; Gibco, 10099-141, USA), 0.23 mmol/L sodium pyruvate (Hyclone, SH40003-12), 100 IU/ml of penicillin G, and 100 mg/ml of streptomycin sulfate (Hyclone, SV30010). Aliquots of 30 µM ZEA were introduced during in vitro culture to establish the ZEA treatment group, the dose of ZEA was based on the study by Zhang et al. [39]. Following the previous laboratory protocol, the siRNA of Hdac2 was transfected to silence gene expression [6, 33]. Reagents used for transfection included: Lipofectamine™ 3000 Transfection Reagent (Lipo 3000, Thermo Fisher Scientific, L3000008, Germany) and 20 µmol/L si-Hdac2 (GenePharma, Shanghai, China; Table S1). The ovaries were cultured in a 37°C, 5% CO2 incubator for three days. The medium was changed for the first time 8 h after transfection, and again one day later.
Immunostaining of ovarian sections
The ovaries were fixed in 4% PFA for 8 h, washed with flowing water, dehydrated, and embedded in paraffin. Then sections were prepared with a thickness of 5 µm. Before staining, the histological sections underwent xylene dewaxing, gradient rehydration, and antigen retrieval. Subsequently, histological sections were used for immunofluorescence (IF) and immunohistochemistry (IHC). For IF, after antigen retrieval and natural cooling, the blocking buffer (containing 10% goat serum diluted and 3% BSA) was used to block the histological sections at room temperature for 45 min; they were subsequently incubated overnight with the primary antibody (Table S2) at 4 ℃. Incubation with the secondary antibody (Table S2) took place at 37°C for 40 min in a dark room the next morning. Hoechst 33342 (Beyotime, C1022, Shanghai, China) or PI (Solarbio, P8080) was used for 3 min at 37°C to stain the nuclei. Finally, the slides were sealed with an anti-fluorescence attenuation quenching agent, before observation of sections under a fluorescence microscope (Olympus, BX51, Japan). For IHC, the same procedure took place as given above; however, before the blocking step, 3% H2O2 was used to treat samples for 10 min. After incubation with primary antibody, each slide was painted with horseradish peroxidase (HRP)–conjugated goat anti-rabbit IgG (Beyotime, A0258) for 45 min at 37 ℃. Subsequently, DAB peroxidase substrate (ZSGB‐BIO, ZLI‐9017, Beijing, China) was used to create a chromogenic reaction, the cell nucleus was stained with hematoxylin, and finally after dehydration with alcohol and permeabilization with xylene, the histological sections were sealed with neutral resin and observed under a BX51 microscope.
Western blotting
The collected ovarian samples were taken from the -80 ℃ refrigerator. The appropriate amount of RIPA (Beyotime, P0013C) was added to lyse ovarian tissues on ice for 20 min; SDS-PAGE Sample Loading Buffer (5×, Beyotime, P0015L) was supplemented and the sample was boiled for 5 min to denature the protein. The 8–15% SDS-PAGE gel electrophoresis was applied according to the molecular weight of the target protein at 100 V constant voltage for 90–130 min. Then the target proteins were transferred onto a polyvinylidene fluoride membrane (PVDF; Millipore, ISEQ00010, USA) at a constant current of 300 mA for 120 min. Subsequently, the protein bands were blocked in TBST solution containing 5% BSA for 4 h on a low temperature shaker. Subsequently, the protein bands were incubated with the primary antibody (Table S2) overnight at 4°C and secondary antibody at room temperature for 75 min. After rinsing in TBST, a BeyoECL Plus kit (Beyotime, P0018) was used for chemiluminescence. AlphaView SA software was used to perform the gray analysis of protein bands.
Acetyl-CoA measurement
A Mouse Acetyl Coenzyme A (ACA) ELISA Kit (JM-1689M2, Jingmei Biological, Jiangsu, China) was used to detect ACA levels. Accordingly, 25 mg of fresh mouse ovaries were collected and lysed in PBS for 30 min. Following the standard requirements of the testing kit, the enzyme-labeled reagents were incubated at 37°C for 60 min, and the treatment of color-developing solution was performed for 15 min under dark conditions. Finally, the stop solution was added to terminate the reaction, and the absorbance value at a wavelength of 450 nm was obtained with a microplate reader within 15 min, which was used for the calculation of ACA content.
Citrate measurement
A Citric acid (CA) content detection kit (Solarbio, BC2150) was used to detect the level of CA. For each test, 25 mg of ovarian tissue was prepared, and the reagents were used in strict accordance with the kit instructions. After standing at room temperature for 30 min, a microplate reader was used to detect absorbance at the wavelength of 545 nm.
Transmission Electron Microscopy (TEM)
Fresh ovaries were fixed overnight with 2.5% glutaraldehyde at 4 ℃. According to standard TEM procedures, the samples were embedded in resin [41]. Serial sectioning with an EM UC7 ultramicrotome (50 nm) was conducted, and the ultrathin sections were stained with lead citrate and uranyl acetate for follow-up observation with HT7700 TEM (Hitachi, Tokyo, Japan).
Detection of mitochondrial membrane potential
Mitochondrial membrane potential of the fresh ovaries was detected using a JC-1 detection kit (Beyotime, C2006). Using trypsin (Sangon, A003702, Shanghai, China) and collagenase (Sigma, C5138, 9:1), the ovaries were digested into single cells at 37°C (30 min), and the samples were incubated with a JC-1 staining working solution at 37°C (20 min). After washing with a JC-1 staining buffer, the samples were diluted and tested for flow cytometry (BD Calibur, CA, USA). Finally, results were analyzed with FlowJo_v.10 software.
Real-Time quantitative PCR (RT-qPCR)
Total RNA was obtained from mouse ovaries with a SPARKeasy Improved Tissue/cell RNA kit (Sparkjade, AC0202, Shandong, China). The extracted RNA was subjected to reverse transcription to synthesize cDNA with a SPARKscript II RT plus kit (With gDNA Eraser) (Sparkjade, AG0304). RT-qPCR was performed using SYBR Premix Ex Taq™ II (Vazyme, Q711-02, Nanjing, China) with a Roche 480 (Roche, Germany) Light Cycler RT-qPCR instrument. All primers are listed in Table S1.
Statistical analysis
All experiments had at least three repetitions between groups. The data were presented as mean ± SD, and the significance of differences between groups was analyzed using the t-test or ANOVA and Graph Pad Prism 8.0 software. P < 0.05 (*) represents a significant difference, and P < 0.01 (**) indicates a remarkably significant difference.
{kind=link}
{kind=link}
{kind=link}
{kind=link}