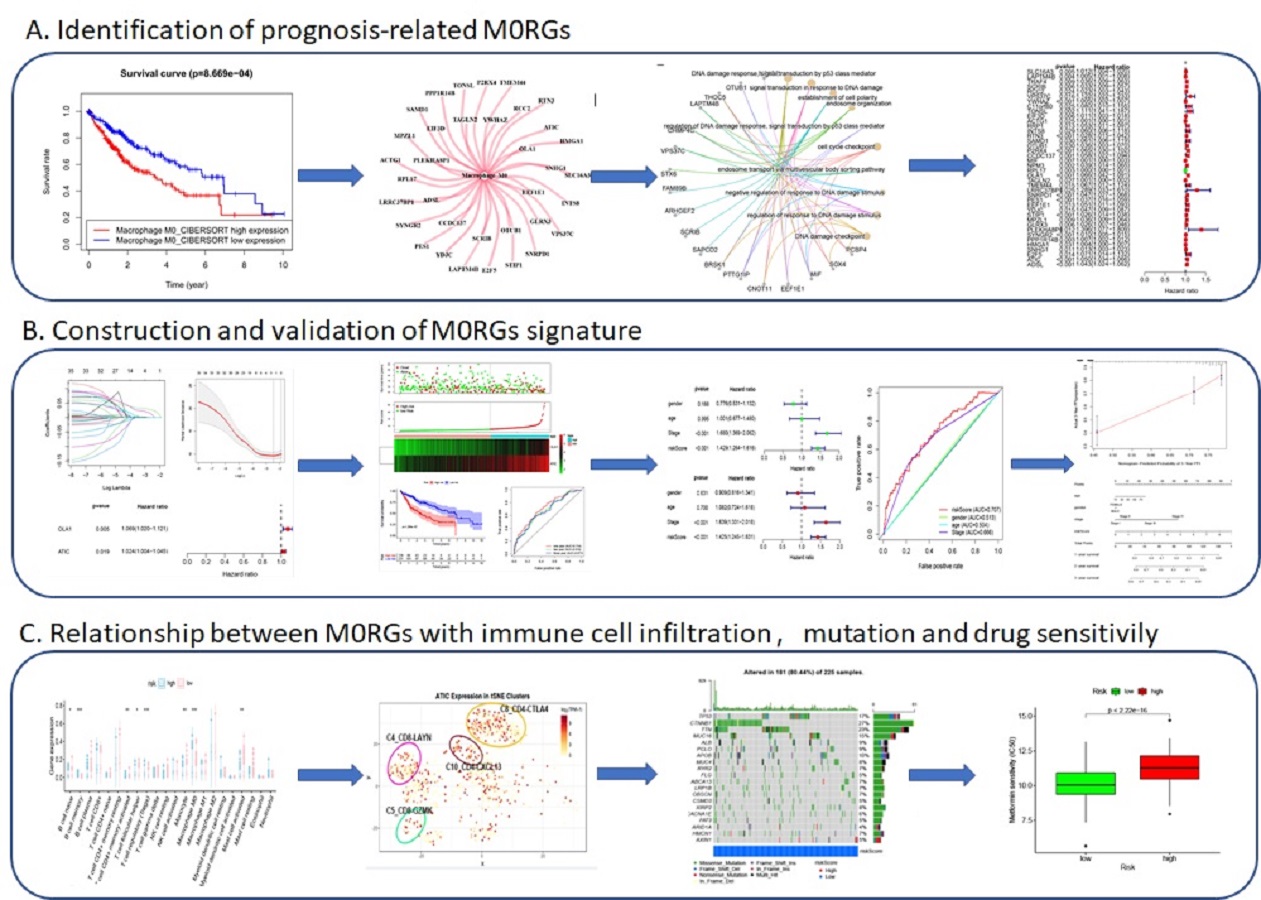
Our comprehensive integrated analysis of M0RGs in HCC enhances the understanding of the molecular events relevant to HCC progression and treatment. The bioinformatics tools used in the current study have facilitated efficient prediction of the composition and changes in the TIME of HCC. The robust statistical power provided by relatively large sample sizes in TCGA and ICGC databases enabled the identification and validation of an M0RG prognostic signature. This is the first M0 macrophage-related risk score model for HCC; the model exhibited a good potential for the evaluation of HCC prognosis and the selection of a therapeutic strategy for HCC. Systematic analysis revealed that high risk scores were associated with a poor prognosis, immune infiltration, and gene mutations, and multivariate analysis confirmed that the risk model was an independent prognostic factor for patients with HCC.
Our results showed that the risk model was positively correlated with CTLA4, PD-L1, and TIM-3 expression, suggesting a potential role of the risk model in evaluating the efficacy of ICI therapy. The liver is the largest immune organ in the human body. Carcinogenic factors, such as persistent hepatitis B and C viral infections [16, 17], can compromise the immune defense or balance, rendering the immune cells unable to remove carcinogens [18, 19]. In early stages of tumor initiation, immune suppression decreases immune surveillance [20]. Thus, ICIs, such as PD-1/PD-L1 inhibitors, have become a promising treatment for HCC as they activate and restore immune functions for the optimal ablation of tumor cells [21–23]. Identifying a predictive model is of great importance for improving HCC immune therapy. We identified the associations between the risk model and drug sensitivity in HCC. A high risk score was associated with lower IC50 values of several drugs, which indicated that this model could be a predictor for drug sensitivity in HCC as a clinical reference. For example, the potential antitumor effects of metformin can be further investigated using the “new uses of old drugs” strategy for drug repositioning.
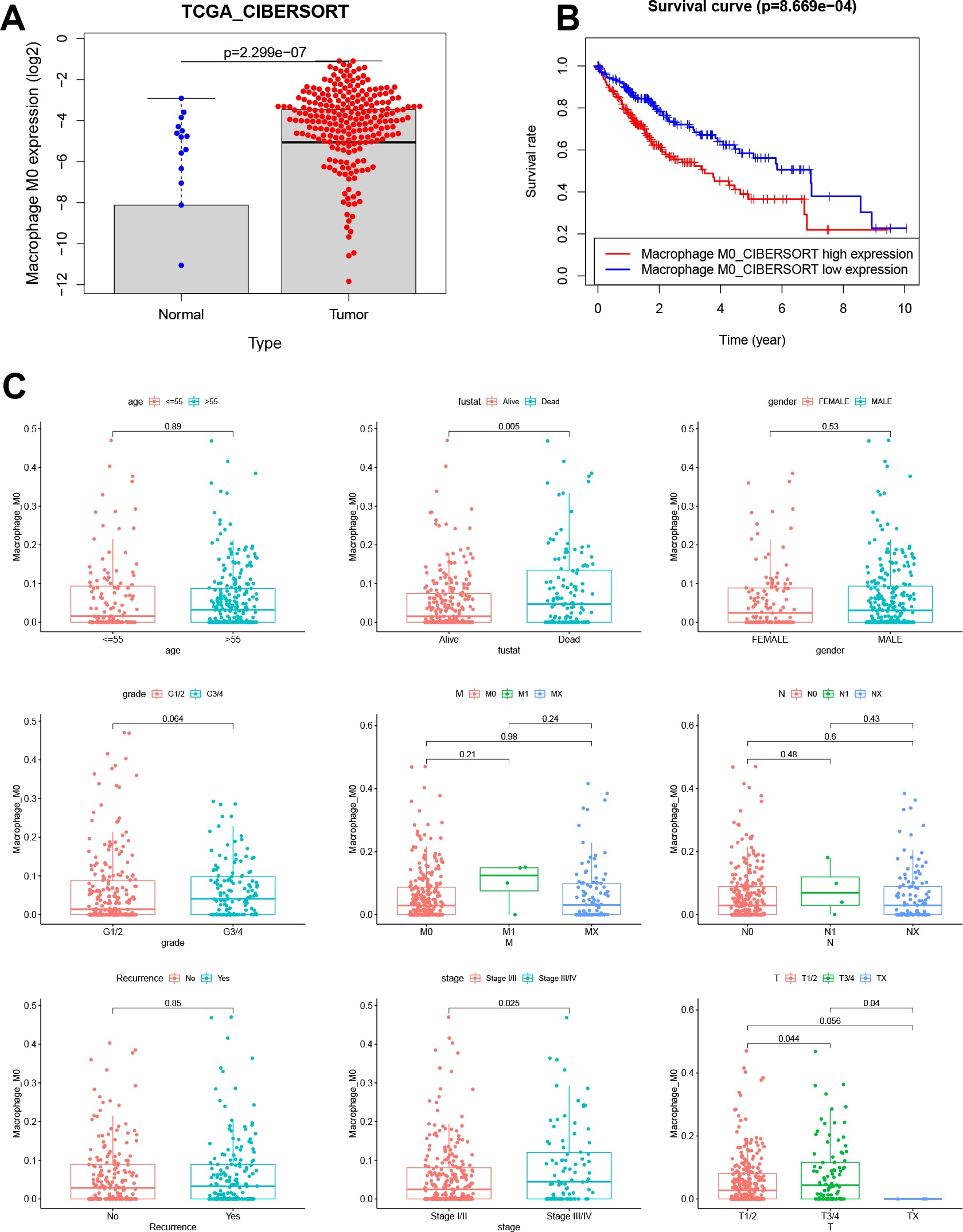
The significance of our study lies not only in the revelation of the composition of infiltrating immune cells in HCC but also in the demonstration of a systematic association of the M0 phenotype and gene clusters with genomic characteristics and clinical features. To this end, we identified biomarkers for potential clinical application. These biomarkers were further used to construct a risk model to predict the prognosis of patients with HCC. Analysis of TCGA datasets revealed that M0 macrophages and relevant genes were unfavorable factors that correlated with clinical features and prognosis of HCC. These results contrasted, to some extent, with previous findings, which suggested that the differentiation of polarized M1 or M2 macrophages was associated with functional properties of tumors [24, 25]. The canonical M1 versus M2 dichotomy has been challenged by recent evidence supporting abundant differentiation of nonpolarized M0 macrophages, rather than that of M1 or M2 macrophages, in tumors [11, 12]. M0 macrophages are defined as undifferentiated macrophages with the potential to polarize into specific macrophage subtypes. Different subtypes of liver macrophages, especially Kupffer cells and TAMs, exhibit diverse ontogeny, differentiation, and function [26, 27]. TAMs have been significantly implicated in HCC initiation, progression, immune evasion, invasion, angiogenesis, and metastasis, as well as in response to therapy [28]. Liver macrophages exhibit highly variable phenotypes that are modulated by signals derived from the liver microenvironment. M1 and M2 macrophages coexist in the tumor microenvironment of various cancers, which may be because of a continuous, rather than isolated, process of M0 macrophage polarization into M1 and M2 macrophages [29]. Based on our findings, it is hypothesized that the infiltration and differentiation of TAMs in the liver are possibly stimulated in response to carcinogenic factors, thus promoting chronic inflammation, suppressing immunity, and leading to HCC progression.
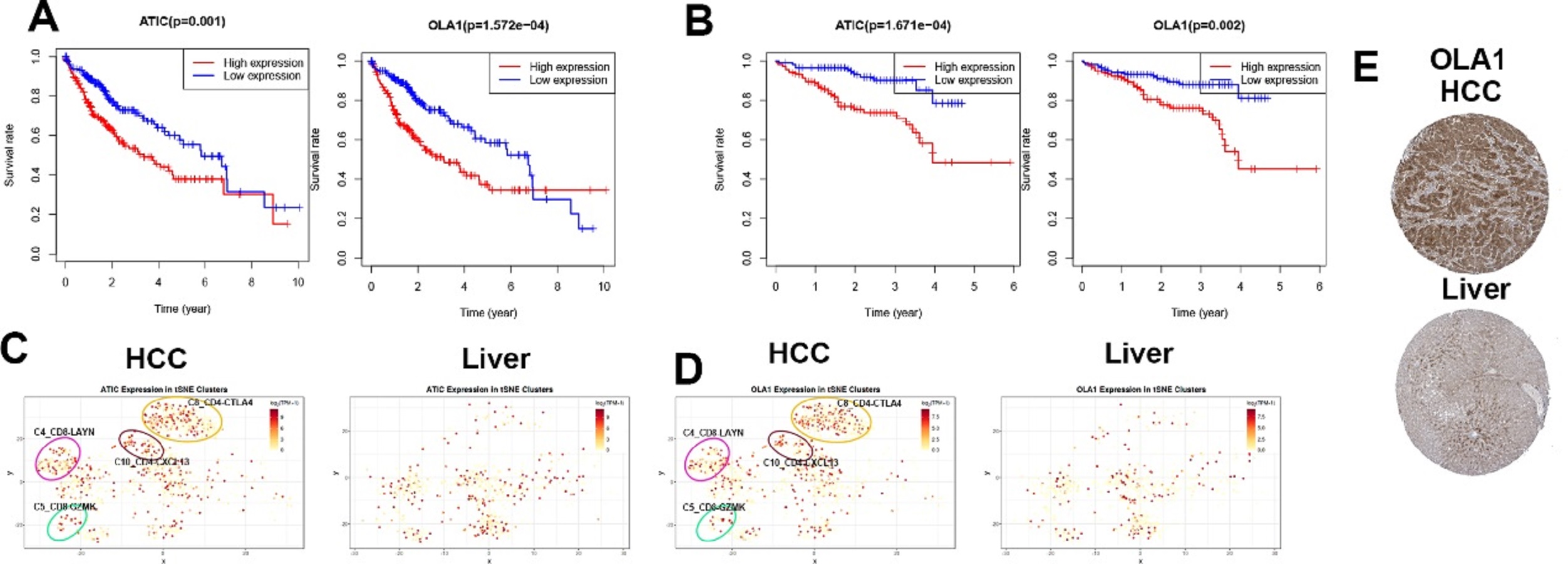
Single-cell analysis of infiltrating immune cells allows in-depth understanding of the landscape of these cells in the highly complicated tumor microenvironment. Recently, single-cell transcriptome technology has been applied to cancerous and immune cells from patients with HCC, resulting in the identification of 11 T cell subsets based on their molecular and functional properties, which delineate their developmental trajectory [30]. In the present study, we analyzed the expression of M0RGs using single-cell sequencing data, and the results revealed that two M0RGs, ATIC and OLA1, were expressed more abundantly in specific subgroups of T cells with signature markers, such as the CD4-CTLA4, CD8-LAYN, CD8-GZMK, and CD4-CXCL13 bundles of HCC tissues. Based on these results, we can propose two scientific hypotheses. First, these specific subgroups might be activated in the HCC microenvironment. The status of T cell infiltration and their characteristics are usually associated with different prognostic outcomes [31]. Several studies have also revealed the association of LAYN, CTLA4, and GZMK expression with tumor-infiltrating exhausted CD8+ T cells and a poor prognosis [30]. Therefore, inhibiting these specific cells might be another strategy for cancer immunotherapy. Second, there may be a crosstalk between macrophages and T cells in the HCC microenvironment, which plays a role in influencing HCC progression and therapeutic efficacy. The polarization and function of HCC-associated macrophages are possibly regulated via these specific subgroups of T cells, which still requires further elucidation.
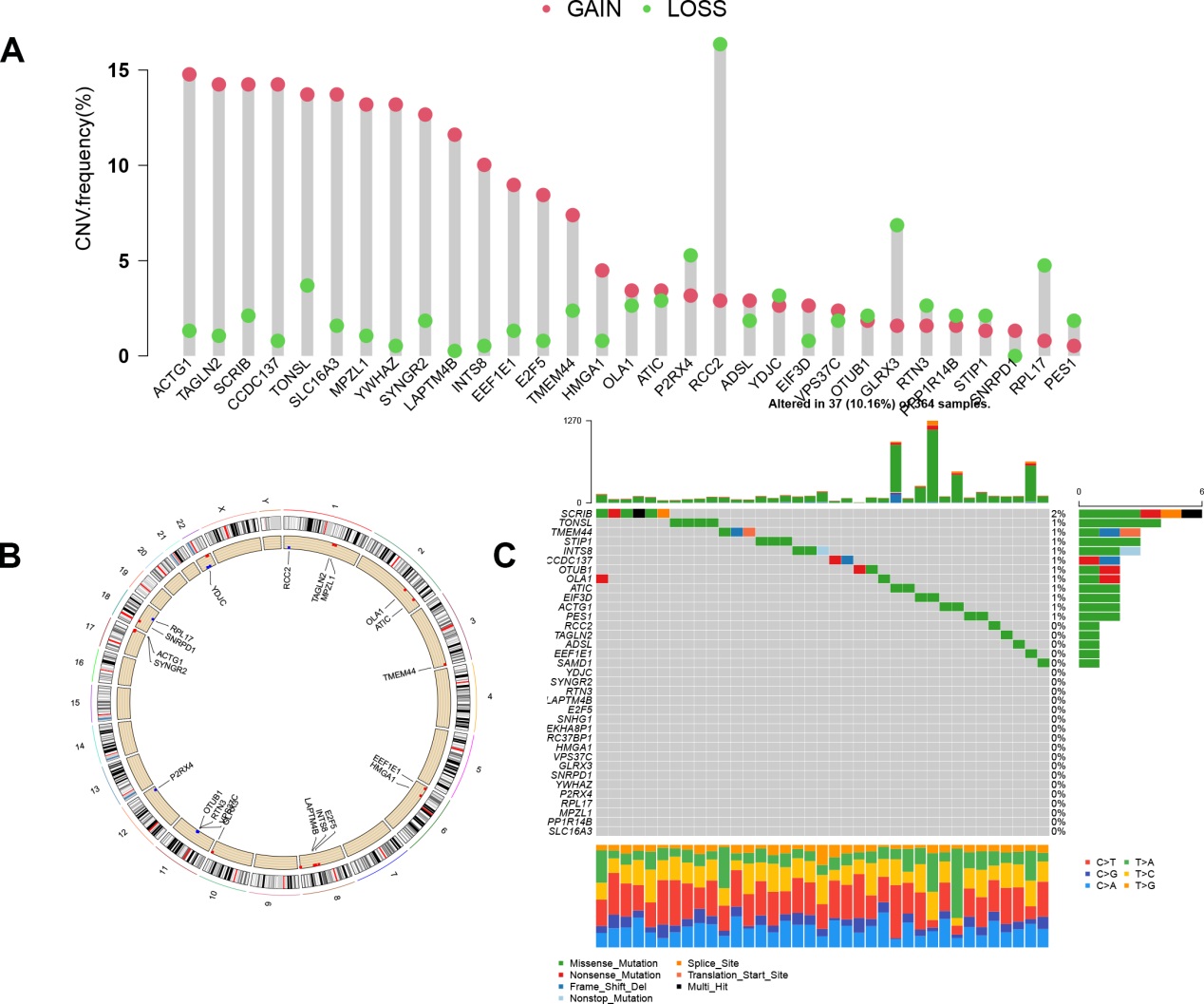
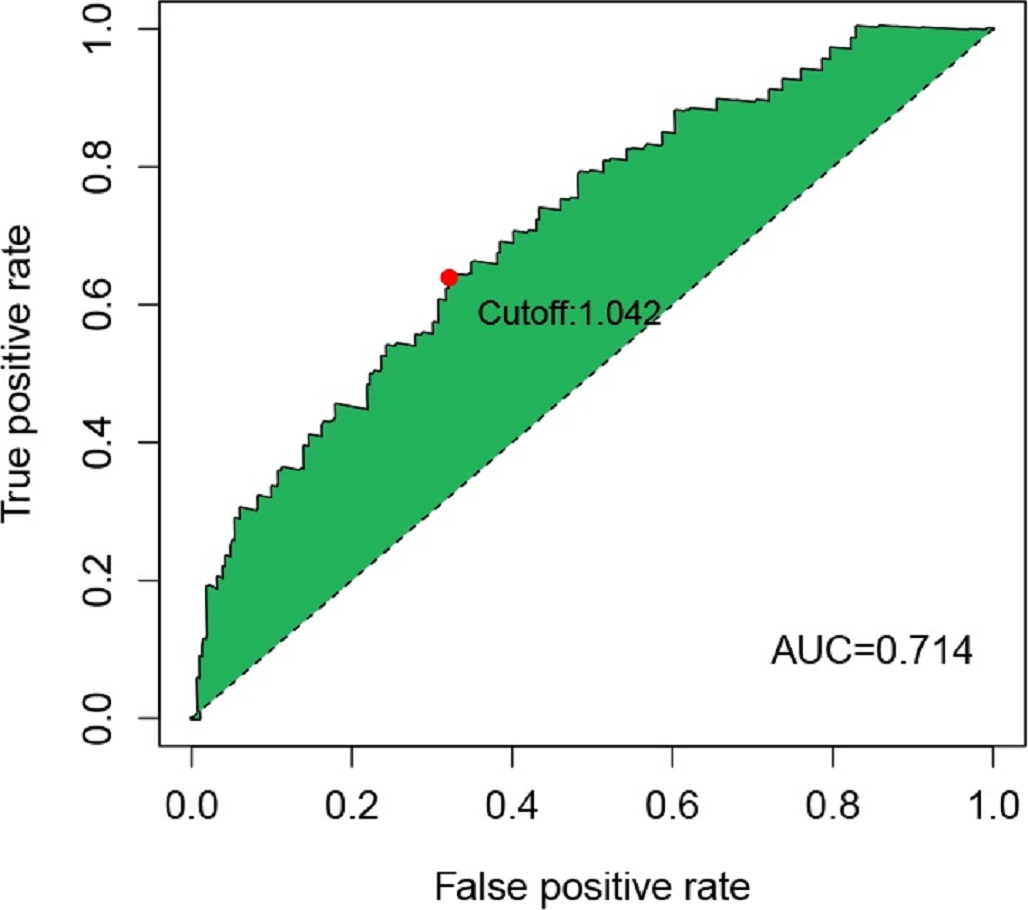
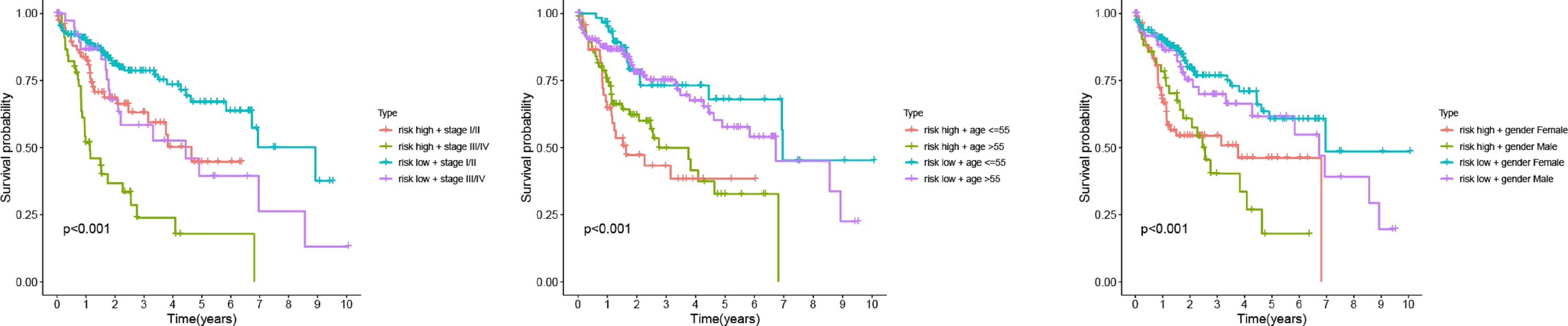
To our knowledge, this is the first study to construct a risk model based on M0RGs in HCC; the model showed that a low risk score reflected a good prognosis, whereas a high risk score indicated a poor prognosis, suggesting that the risk model is a robust prognostic biomarker. Further analysis revealed that cancer- and immune-related signaling pathways were enriched in the high-risk group. B memory cells, Tregs, and M0 macrophages were positively associated with the risk score. These results are in agreement with the prevailing knowledge that pathological division of cells is the basis of tumorigenesis and that immune tolerance can facilitate tumor development [29, 32]. Additionally, we observed that our risk model was associated with known somatic mutations in TP53. These alterations in a somatic gene may inactivate tumor suppressor genes and cause mutations in protooncogenes, resulting in tumorigenesis [33]. Therefore, our study contributes to the identification of immunotherapeutic targets to inhibit the pathways involved in tumorigenesis.
The major limitation of our study is the lack of biological validation of immune cell infiltration in vitro and in vivo in HCC tissues because of a delayed arrival of antibodies due to the coronavirus disease pandemic. Unlike other studies on immune infiltration in HCC [34], our analysis mainly focused on M0 macrophages using a large number of HCC samples from public databases. In addition to the function of immune cells in the TIME, we comprehensively mapped the landscape of interactions involving M0 macrophage-associated immune cells, genes, and clinicopathological features. Moreover, we confirmed that the prognostic value of the risk model was superior to that of other clinical signatures. This precise and simple model of two M0RGs will contribute to evaluating the prognosis of and treatment efficacy in HCC. We also revealed promising immune-based candidate biomarkers for the diagnosis, prognosis, and therapy of HCC.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}