Synthesis and characterisation of HEAs
Quaternary, quinary, and nonary alloys were synthesised by conventional arc melting. Mother metal ingots were melted uniformly to obtain a 20-g button ingot of quaternary (Nb, Zr, Mo, and Ti; denoted as 4eHEA), quinary (Cr, Mn, Fe, Co, and Ni; denoted as 5eHEA), or nonary (Ti, Cr, Mn, Fe, Co, Ni, Zr, Nb, and Mo; denoted as 9eHEA) alloy (Fig. 1a). The crystal structures suggest that 9eHEA contains new phases unlike those seen in 4eHEA and 5eHEA (Fig. 1b). X‐ray fluorescence (XRF) spectroscopy results show that all HEAs contained different metals at almost the same molar ratios (Supplementary Table 1). The button ingot was mechanically crushed by ball milling to reach a particle size of 500–1000 nm (Supplementary Fig. S1). The specific surface areas measured by nitrogen adsorption/desorption method based on the Brunauer–Emmett–Teller (BET) theory26 are 43.1, 2.7, and 4.7 m2g−1 for 4eHEA, 5eHEA, and 9eHEA, respectively (Supplementary Fig. S2). Dark-field scanning transmission electron microscopy (DF-STEM) was used to examine the mechanically crushed 9eHEA particles (Fig. 1c). The high-resolution DF-STEM image revealed an amorphous-like lattice with high and low intensity distributions at the atomic scale (Fig. 1d and Supplementary Fig. S3), indicating an absence of obvious segregation in any of the elements. In addition, the elemental maps indicate that the nine elements were uniformly distributed in the alloys, and therefore there was no obvious phase separation at the atomic scale on the crushed particles. We further investigated the chemical binding states of HEAs after ball milling through X-ray photoelectron spectroscopy (XPS). XPS can detect metallic states of elements in HEA with partial oxidation under air conditions (Supplementary Figs. S4–S7). For electrochemical tests, the milled HEAs were mixed with commercially available carbon black (CB) as dispersing supports (Supplementary Figs. S2 and S8).
Corrosion behaviours of HEAs
The corrosion behaviours of the HEAs were studied in a N2-saturated 0.5 M aqueous H2SO4 electrolyte at 25 °C (Fig. 2a and Supplementary Fig. S9). The anodic polarisation curve from −0.55 to 2.2 V (vs. reversible hydrogen electrode (RHE)) and the cathodic polarisation curve from 2.2 to −0.55 V (vs. RHE) for the HEAs demonstrate a single cross potential (Ecoor). From the anodic polarisation curves of 9eHEA, the corrosion current density (Icoor) at Ecoor was estimated27 as 1.26 μA cm−2 at 0.11 V. This Ecoor value is close to that of bulk Cu (0.22 V)28. The Icoor value is higher than that of bulk Pt29, but lower than those of Pt nanoparticles30, Pt-based alloy nanoparticles31, bulk non-noble metals (Ni, Cu, and Fe)28,32,33, 304 stainless steel34, Ni phosphide alloys32, high entropy alloys34–36, 4eHEA, and 5eHEA at similar conditions (Fig. 2b and Supplementary Table 2). Moreover, the polarisation curves of 9eHEA at 2.20 V (vs. RHE) reveal no obvious hysteresis (Supplementary Fig. S9), which indicates that this HEA has high corrosion resistance without pitting-type corrosion37.
EC-XPS measurements of 9eHEA
The surface states and corrosion behaviours of 9eHEA were further investigated by EC-XPS in an N2-saturated 2.5 mM aqueous H2SO4 electrolyte (Figs. 2c–f, Supplementary Figs. S7 and S10, and Supplementary Tables 3 and 4). The linear sweep voltammograms were obtained from 0.14 (open circuit potential, (OCP)) to −0.30 V for HER processes and from 0.14 (OCP) to 1.9 V for OER processes. There were no significant differences in the EC-XPS data between the OCP and HER state, indicating that the metallic state of HEA was preserved during the reduction processes (Figs. 2d–f). In the OER processes at OCP, metallic Ti, Cr, Zr, Nb, and Mo were partially oxidised to TiO2, Cr(OH)3, ZrO2, Nb2O5, and MoO2 as oxide layers, respectively. Mn, Fe, Co, and Ni were mostly maintained despite some partial dissolution. At the 1st oxidation stage (0.92 V), Ti, Cr, Mn, Zr, and Nb were further oxidised. Mo was oxidised to MoO2 and MoO3. Mn, Fe, Co, and Ni were partially oxidised to Mn2O3, Fe2O3/Fe3O4, Co(OH)2, and Ni(OH)2, respectively. At the 2nd oxidation stage (1.65 V), Cr, Co, and Ni showed potential-dependent oxidation, which turned Cr(OH)3, Co(OH)2, and Ni(OH)2 to CrO3, CoO/CoO2, and NiO/NiO2, respectively. At the OER stage (1.90 V), all elements on the surface were completely oxidised. After OER, the OCP was negatively shifted to 0.89 V, and so the OER process was irreversible. This could be attributed to metal leaching from 9eHEA during the OER processes and the redox reactions of Cr, Co, and Ni. Note that the shift in binding energy of Ti, Zr, Nb, and Mo is proportional to the applied potential between OCP and 1.90 V (Supplementary Fig. S10), which indicates that their chemical states (i.e. oxidised state) are potential-independent. Hence, their oxides work as passivation layers. These results during the OER processes suggest that the nine metals play three different roles: (i) passivation (Ti, Zr, Nb, and Mo), (ii) redox-active (Co, Ni, and Cr), and (iii) others (Mn and Fe).
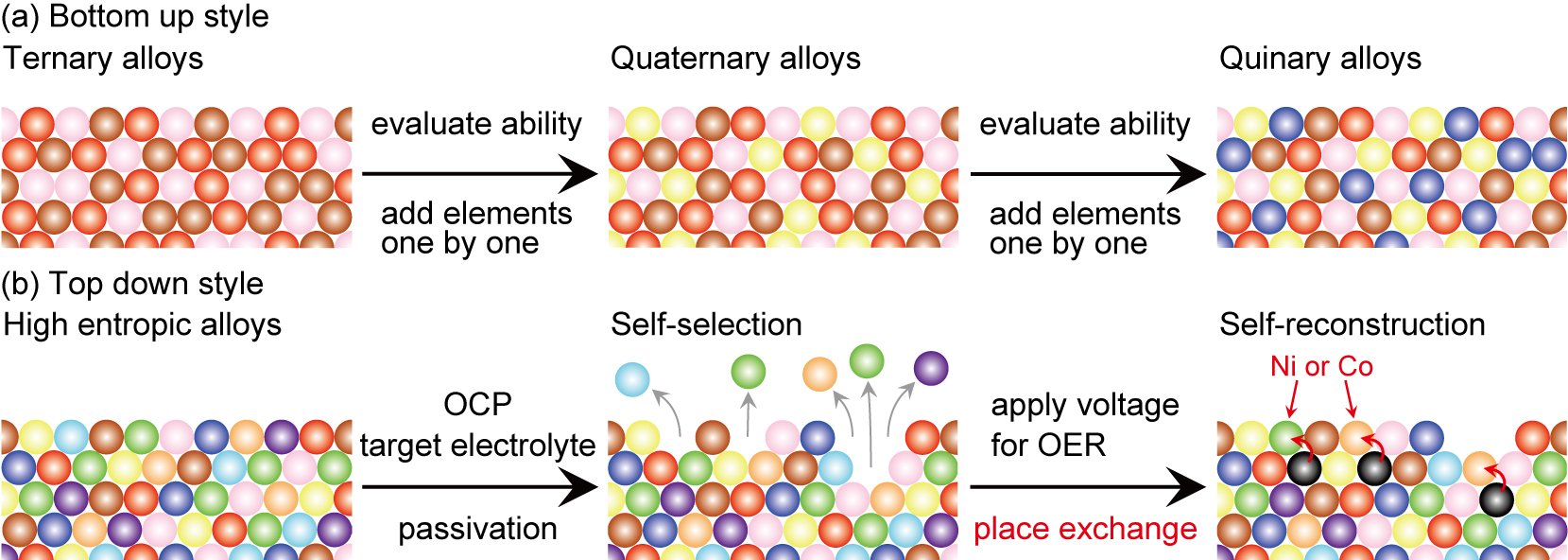
Depth analysis of each element was carried out using angle-resolved EC-XPS (Supplementary Figs. S7 and S11 and Supplementary Table S4). XPS data at the HER stage are similar to that at OCP, indicating that all elements had identical distribution. In contrast, XPS data at the OER stage indicate that Ti, Cr, Mn, Fe, Zr, Nb, and Mo were distributed uniformly along the depth direction, while Co and Ni showed angle-dependent distributions. Considering the higher concentrations of Co and Ni atoms on the surface at 1.90 V than at other potentials, these two elements were segregated only at the OER stage due to the place exchange38,39 with oxygen species (Scheme 1). After the OER stage, Co and Ni penetrated the underlaying passivation layers. Thus, the OER activity was enhanced by the self-reconstruction of Co and Ni (i.e., segregation on the surface) that only occurs during the OER stage.
PEM-type water electrolysis
After confirming the high corrosion resistance of our HEAs, we employed them as anode or cathode catalysts in a membrane-electrode assembly (MEA) to verify the feasibility of a single-cell (4.0 cm2) PEM-type electrolyser (Supplementary Fig. S12). To understand the catalytic activity of each HEA on individual electrodes, we prepared three types of MEAs (Supplementary Fig. S13): commercially available Pt/C cathode catalyst + HEA anode catalyst (abbreviated as Pt/C-HEA), HEA cathode catalyst + commercially available IrO2 anode catalyst (abbreviated as HEA-IrO2), as well as Pt/C cathode catalyst + IrO2 anode catalyst (abbreviated as Pt/C-IrO2) for benchmarking. Before the test, we optimised the ball-milling conditions and the catalyst loading (Supplementary Fig. S14). To fairly compare the catalytic activity of different HEAs, the I–V curves obtained using 4eHEA, 5eHEA, and 9eHEA as anode and cathode catalysts were normalised by the respective BET surface area of HEAs (Figs. 3a–3b and Supplementary Fig. S15). When HEA was used as cathode or anode, 9eHEA-IrO2 and Pt/C-9eHEA showed higher performances than those using 4eHEA or 5eHEA. The cell voltage of 9eHEA at 1.0 A cm−2 current density normalised by the electrode surface area (4.0 cm2) show 0.30 V (cathode) and 0.56 V (anode) higher than that of Pt/C-IrO2 (1.58 V) (Fig. 3c). Their performances are comparable with reported noble metal catalysts and noble/non-noble alloy metal catalysts in the PEM electrolyser (Supplementary Table S5). The 1.88 V (78.7%) and 2.14 V (69.2%) cell voltages of 9eHEA at 1.0 A cm−2 bring 15.0% and 24.5% energy conversion efficiency loss of the electrolyser7 in comparison to the Pt/C-IrO2 case (93.7%). Moreover, from the electrical impedance data, their Ohmic resistance and charge transfer resistance are close to those of Pt/C-IrO2 (Supplementary Fig. S16).
Next, the cycling stability and durability of HEA catalysts were investigated (Figs. 3c and 3d). After 1,000 cycles, Pt/C-9eHEA showed no degradation at 5.0 A cm−2, while 9eHEA-IrO2 showed 5.4% reduction. Chronoamperometry (CP) tests of 9eHEA at a current density of 1.0 A cm−2 for 100 h resulted in a current density reduction of 13% (cathode) and 0% (anode). After the CP test, the morphology and chemical state of catalysts on the MEA were investigated. Cross-sectional SEM images of MEA and XRD spectra demonstrate that the original microstructures were well preserved (Supplementary Figs. S17 and S18). DF-STEM and elemental mapping of 9eHEA anode catalysts revealed a homogeneous distribution without obvious pitting corrosion (Supplementary Fig. S19). XPS data of 9eHEA in the cathode and anode confirmed the metallic state and heavily oxidised state, respectively (Supplementary Figs. S20 and S21). After the CP test, the alloy’s composition remained almost unchanged (except for Nb as Nb2O5 at the OER), which indicate that the surface structures are stable after the self-selection, and there was no Ir contaminations from the system (Supplementary Fig. S22). For comparison, the cycling stability of 4eHEA and 5eHEA was similarly investigated (Supplementary Fig. S23). Pt/C-4eHEA showed no degradation but low performance, whereas Pt/C-5eHEA showed high performance as well as degradation (Figs. 3a and 3b). These results indicate that the combination of Ti, Zr, Nb, and Mo (i.e., 4eHEA) yields good passivation performance, while that of Cr, Mn, Fe, Co, and Ni (i.e., 5eHEA) gives good catalytic performance.
Computational study of the catalytic mechanism
To understand the catalytic activities of each element in the HEA and obtain deep insight from the EC-XPS results, we modelled the structures of 9eHEA (Fig. 4a) and estimated the Gibbs free energy for HER and OER processes by high-throughput DFT calculations, with the aid of a machine-learning force field (MLFF) generated from DFT-molecular dynamics (MD) simulation. We generated 49 slab models, whose surface areas were around 1.5 nm × 1.5 nm. The computational details and typical adsorption structures are given in Supplementary Information and Supplementary Figs. S24–S28. In the case of HER40,41, highly efficient catalysts tend to have their Gibbs free energy for H* adsorption, |ΔGH*|, close to zero. For example, ΔGH* for Pt (ΔGH*Pt) is approximately −0.08 eV. ΔGH* of each element on the 9eHEA surface at 1796 calculation sites can take positive (2.3%) or negative (97.7%) values (Fig. 4b). In particular, Mn (range: −0.0088 to −0.097 eV, 7 sites) and Fe (range: −0.0064 to −0.106 eV, 8 sites) could be active sites as effective as Pt. Note that the nearest neighbour elements to the catalytically active sites with |ΔGH*| < 0.10 eV are Cr and Fe (15/26 sites) (Supplementary Table 6), which means that these elements enhance the catalytic activity of the active sites. Moreover, we calculated the Gibbs free energies of the OER processes42,43 at 270 sites, and focused on the |ΔGO*| (strong adsorption of O* intermediate on the catalytic site) under an applied potential of 1.23 V (vs. RHE) as the rate-determining step in the OER processes (Fig. 3c). Fe, Co, and Ni showed low adsorption energies of ΔGO* = −2.36, −1.98, and −1.00 eV, respectively, in agreement with the experimental findings. Note that for the catalytically active sites in the system with the largest |ΔG| < 3.0 eV, 71% of their nearest neighbour elements are Fe, Mn, or Cr (27/38 sites, Supplementary Table 7). Therefore, these three elements promote OER processes on the active sites. The interplay of active sites and their nearest neighbour elements is important for enhancing the catalytic activities for HER and OER. Furthermore, we simulated the OER processes on an initially oxidised surface to reflect the real OER situation by using the model with the segregated Ni suggested by EC-XPS (Supplementary Fig. S11). The pre-oxidised surface reduced the activation energy by 0.99 eV (from −2.00 eV to −1.02 eV, Fig. 3f). Thus, the oxidation state near catalytically active sites is also crucial for enhancing the OER activity and this finding arises as a consequence of the self-selection/reconstruction (Scheme 1).
{kind=link}