Animals
Male adult Sprague-Dawley (SD) rats were used for detection. All animals received humane care, and study protocols complied with the Laboratory animal—Guideline for ethical review of animal welfare (GB/T 35892—2018) and were approved by the Medical Ethics Committee of Lanzhou University (jcyxy20190302). This study also conformed with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines and Replacement, Refinement and Reduction of Animals in Research (NC3Rs).
Sample collection and contamination avoidance
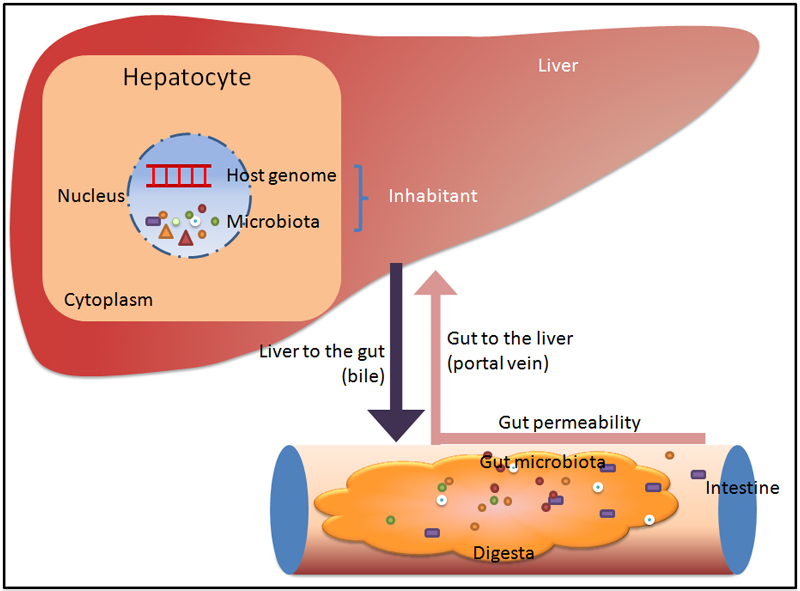
Animals were anesthetized with pentobarbital sodium (5 mg/100 g body mass) followed by three skin sterilizations using 75% ethanol and iodophor. To avoid environmental contamination, all operations were conducted under a laminar-flow hood following aseptic-surgery protocols, including the use of a surgical towel, and avoided contact between skin and subcutaneous tissue. After the skin was opened, we discarded the original set of all surgical instruments and used another set. To eliminate possible blood contamination, animals were sacrificed by portal-vein blood drainage. To prevent possible fecal contamination, we began by removing liver tissues, then those of the ileum (small intestine [SI]) and colon (large intestine [LI]). Subsequently, SI and LI contents were pushed out from the outside of the gut using another set of tweezers. All samples were stored at −80°C after a quick freeze in liquid nitrogen. To avoid possible contamination during 16S rRNA gene analysis, we transported samples on dry ice to a sequencing company (BMK Co., Beijing, China), which provided an integrated service in DNA extraction, polymerase chain reaction (PCR) amplification, and sequencing via procedures that strictly avoided contamination.
Bacteria DNA extraction
We extracted total DNA from frozen liver and fecal samples (0.25–0.5 g) using a Magnetic Soil and Stool DNA Kit (#DP812; TianGen Corp., Beijing, China, https://www.tiangen.com/) per the kit protocol. All procedures were performed with sterile and disposable materials to avoid cross-contamination, including the beads.
16S PCR and sequencing library construction
We used two-round tailed PCR with the barcode at the end of primer for 16S amplification and sequencing. In the first round, the 16S rRNA gene’s variable regions 3–4 (V3–V4) of interest was amplified with an initial heating step of 95ºC for 5 min, followed by 25 cycles of 30 s at 95ºC, 30 s at 50ºC, 40 s at 72ºC and a final extension step of 72ºC for 7 min. The bacterial primer was as follows: 338F: 5'-ACTCCTACGGGAGGCAGCA-3'; 806R: 5'-GGACTACHVGGGTWTCTAAT-3' [10]. The reaction system was as follows: bacterial-genome DNA, 50.0 ng ± 20%; primers (338F and 806R, 10 μM; Synbio Technologies, Suzhou, China), 0.3 μl each; KOD FX Neo Buffer (KFX-201S; TOYOBO; Biolink Biotechnology, Beijing, China, http://www.bjbiolink.com/), 5.0 μl; deoxyribose nucleotide triphosphate (dNTP; 2 mM each), 2.0 μl; KOD FX Neo, 0.2 μl; and double-distilled water (ddH2O) added to 10.0 μl. We performed the PCR reaction using an Applied Biosystems PCR System (Veriti 96-Well 9902; Applied Biosystems, Foster City, CA, USA). PCR products (10-μl system) were purified using VAHTSTM DNA clean beads (Vazyme Corp., Nanjing, China, http://www.vazyme.com/) at a ratio of 1:1, and eluted using 8.0–10.0 μl ddH2O. In the second round of PCR reaction (Solexa PCR), dual-indexed sequences (barcodes) and Illumina adaptors (Illumina, Inc., San Diego, CA, USA) were added to the amplicon. The reaction system was as follows: purified V3–V4 PCR production DNA, 5.0 μl; primers (MPPI-a and MPPI-b, 2.0 μM; Synbio Technologies, Suzhou, China), 2.5 μl each; NEBNext® UltraTM II Q5® Master Mix (New England Biolabs, Ipswich, MA, USA) (M0544L; Biolink Biotechnology, Beijing, China) 10.0 μl. The PCR reaction was performed at 98°C for 30 s, with 10 cycles of 98°C for 10 s, 65°C for 30 s, 72°C for 30 s, and an extension at 72°C for 5 min.
We detected the products using 1.8% agar gel (120 V, 40 min) and qualified them by analyzing the gel images via ImageJ software (National Institutes of Health, Bethesda, MD, USA). Next, we mixed 150 ng of each sample (samples were 1.5–14.0 μl) and purified it using an E.Z.N.A. Cycle-Pure Kit (Omega Bio-Tek, Inc., Norcross, GA, USA).
Sequencing
After quality testing on a Qsep-400 (BiOptic, Inc., New Taipei City, Taiwan, ROC) and preparation of a flow cell chip, we subjected 500 ng PCR products to paired-end (PE) sequenced on an Illumina HiSeq 2500 platform (Illumina, Inc., San Diego, CA, USA) at Biomarker Technologies Co, Ltd. (Beijing, China) according to standard protocols. The sequencing length was 350–450 bp. Original image data files were transformed into raw data (PE reads) via base calling analysis.
Quality assessment and data processing
According to the overlapping relationship, PE reads were merged with Fast Length Adjustment of SHort reads (FLASH) software v.1.2.7 (Johns Hopkins University Center for Computational Biology, Baltimore, MD, USA; raw tags) [11].We discarded tags with > 6 mismatches. The minimum overlap length was 10 bp, and the maximum mismatch ratio allowed in the overlap region was 0.2 (default). Raw tags with an average quality score < 20 in a 50-bp sliding window were then filtered using Trimmmatic software v.0.33 (USADELLAB.org) [12] and those shorter than 350 bp were removed (clean tags). We further removed possible chimeras using UCHIME v.4.2 (http://drive5.com/usearch/manual/uchime_algo.html) [13] to obtain effective tags.
Operational taxonomic-unit analysis
Effective tags were clustered at a 97% similarity level to obtain OTUs using USEARCH software v.10.0 [14]. We evaluated the α-diversity index of each sample using Mothur software v.1.30 (http://mothur.org/) and the β-diversity index using Quantitative Insights Into Microbial Ecology (QIIME) software v2.2. Degrees of similarity in species diversity between different samples were compared.
Taxonomic analysis
We compared representative OTU sequences against the Silva microbial reference database (release 128; http://www.arb-silva.de) [15]. The classification information of each OTU was obtained by comparison, and the OTU was annotated using RDPClassifier (v2.2; QIIME) [16]. Next, we counted the community composition of each sample at phylum, class, order, family, genus, and species levels. Species richness at different taxonomic levels was assessed using QIIME, and the community structure diagram of each taxonomic level was drawn using R software v3.1.1 (R Foundation for Statistical Computing, Vienna, Austria).
Immunofluorescence assays
Immunofluorescence (IF) staining was performed according to standard staining methods as described, with slight modification [5]. Briefly, we fixed frozen liver tissue sections (4 μm) with 4% paraformaldehyde for 30 min and aged them at 60°C for 10 min, followed by incubation with 0.3% Triton X-100 (#T8200; Solarbio) and 3% bovine serum albumin (BSA; B2064-50G; Sigma Germany, Munich, Germany) for 30 min. Primary antibodies (lipopolysaccharide (LPS) core, monoclonal antibody [mAb] WN1 222-5, #HM6011, 1:400 dilution; lipoteichoic acid (LTA), mAb 55, #HM2048; 1:400 dilution; both from Hycult Biotech, Uden, Netherlands) or phosphate-buffered saline (PBS; #BL601A; BioSharp Life Sciences, Hefei, China) as negative control were applied to slides overnight at 4˚C; secondary antibody (DyLight 488 goat anti-mouse immunoglobulin G [IgG]; Ex/Em = 493/518 nm, #AMJ-AB2004; 1:800 dilution; AmyJet Scientific Inc., Wuhan, China) were added for 1 h at 37°C. We counterstained the slides with 4′,6-diamidino-2-phenylindole (DAPI; 0.5 mg/ml; Ex/Em = 364/454, #BL105A; BioSharp) at room temperature (RT) for 10 min, and then dried the slices in a dark room and mounted them with an anti-fluorescence attenuator mountant. Paraformaldehyde fixed Staphylococcus and Escherichia coli (preserved and kind gifts from Professor Jian Han (Lan Zhou University, China) were used as Gram-positive and Gram-negative controls, respectively. We observed and recorded slides using Nikon-ECLIPSE 80i/DS-Ri2/NIS-ElementsD microscopy (Nikon, Tokyo, Japan).
Equipment and settings
Images were captured with software NIS-Elements D using an Nikon-ECLIPSE 80i upright fluorescence microscope outfitted with a Nikon DS-Ri2 camera, Plan Fluor 40x DIC M N2 objectives (objective numerical aperture: 0.75, refractive index: 1.000). The images were acquisited with 96 dpi in x- and y-axis with 4908 × 3264 pixel dimensions, and the image bit depth was 24.
16S rRNA gene fluorescent in situ hybridization
We detected the 16S rRNA gene in tissues according to the instructions of a EUB338 FISH Probe Kit (#20 μM; FBPC-10; Creative Bioarray, Shirley, NY, USA). Briefly, frozen tissue slides (4 μm) were fixed with 4% paraformaldehyde for 30 min and at 60°C for 10 min, followed by incubation with 0.3% Triton X-100 for 30 min. We then incubated slides in lysozyme at 37°C for 15 min. As a positive control, a bacteria smear was incubated in 0.01 M HCl at RT for 20 min. Slides were fixed with 4% paraformaldehyde for another 15 min at RT, after which we treated them with diethyl pyrocarbonate (DEPC) for 10 min and incubated them with BSA (3%) for 2 h. BSA was then discarded, and fluorescein isothiocyanate (FITC)–labeled probes (EUB338 GCTGCCTCCCGTAGGAGT) and a non-specific complement probe (nEUB338 CGACGGAGGG CATCCTCA; #FBPC-13; Creative Bioarray) were hybridized in a pre-warmed humidified hybridization chamber and incubated overnight at 39˚C (range, 38–42°C). We diluted FISH probe EUB338 or nEUB338 with 35% hybridization buffer (1:100), denatured it at 84°C for 5 min, and then incubated at 37°C for 3 min. After hybridization, we carefully removed the sealing film by soaking the slides in wash solution (WS; 2 × saline-sodium citrate [SSC]/0.1% Tween 20) at RT for 15 min to loosen the coverslips. Slides were then rinsed twice in WS for 15 min each time, immersed in 75% and 100% ethanol for 2 min, and then air-dried for 20 min. We counterstained slides with DAPI antifade solution (using EUB338 FISH Probe Kit) for 10 min and examined them under the Nikon fluorescence microscope.
LPS and LTA western blotting
We separated cytoplasmic from nuclear components using a Minute Cytoplasmic and Nuclear Fractionation Kit (#sc-003; Invent, Beijing, China). The liver tissue was washed with pre-cooled sterile PBS, and 60 mg of fresh (or frozen) soft liver tissue was added to a 1.5-ml sterile microcentrifuge tube. We then added 200 μl of pre-cooled sterile PBS to the same tube and ground tissue with a clean plastic grinding rod for 2–3 min on ice until no solid tissue was visible, after which the frozen liver tissue sample was thawed completely on ice and then ground again. After incubation on ice for 5 min, we carefully transferred the supernatant into another pre-cooled sterile 1.5-ml microcentrifuge tube. Cells were harvested from the suspension by low-speed centrifugation (500 × g) at 4°C for 3 min. We added 200 μl cytoplasmic extraction buffer for every 20 μl of cell volume, vortexed the tube vigorously for 15 s, and incubated it on ice for 15 min. Then, we centrifuged it for 5 min at 16000 × g and 4°C in a microcentrifuge. The supernatant (cytosol fraction) was transferred to a fresh pre-chilled 1.5-ml tube; the pellet was washed in 0.5 ml cold PBS, centrifuged at 8000 × g for 5 min to reduce contamination of cytosolic proteins and frozen at −80°C. We added 100 μl nuclear extraction buffer to the pellet (ratio of cytoplasmic to nuclear extraction buffer, 2:1), vortexed the mixture vigorously for 30 s and incubated the tube on ice for 2 min; this sequence of steps was repeated five times. Immediately afterward, we transferred the nuclear extract to a pre-chilled filter cartridge via a collection tube and centrifuged it at 16000 × g in the microcentrifuge for 30 s at 4°C. We discarded the filter cartridge and stored the nuclear extract at −80°C. The bicinchoninic acid (BCA) method was used to measure protein concentrations. Protein samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE; 12% separating gel), transferred onto a polyvinylidene difluoride (PVDF) membrane (MilliporeSigma, Burlington, MA, USA) , and probed with the above-indicated primary antibodies at 4°C overnight, followed by the appropriate secondary horseradish peroxide (HRP)–conjugated IgG antibody at RT for 1 h. The following antibodies were used: primary, LPS (1:400; HM6011; Hycult), LTA (1:1000; #HM2048; Hycult), Lamin-B1 (1:2000, #ab16048; Abcam, Cambridge, UK), glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:4000; #YM3215; Proteintech, Chicago, IL, USA); secondary, HRP-labeled goat anti-rabbit (1:5000, #RS0002; ImmunoWay Biotechnology Co., Plano, TX, USA) and HRP-labeled goat anti-mouse (1:5000; #RS0001; ImmunoWay). Protein bands were visualized using an electrochemiluminescence (ECL) kit (Super ECL Detection Reagent, Yeasen Biotechnology Co., Ltd., Shanghai, China) and ImageJ software v6.0.
Statistical analysis
All data were expressed as mean ± standard deviation. We conducted all statistical analyses using SPSS software v19.0 (IBM Corp., Armonk, NY, USA). An independent-sample t-test was used for comparisons between two groups, and one-way analysis of variance (ANOVA) was used for mean comparisons between groups. Differences were considered statistically significant at P < 0.05.
{kind=link}