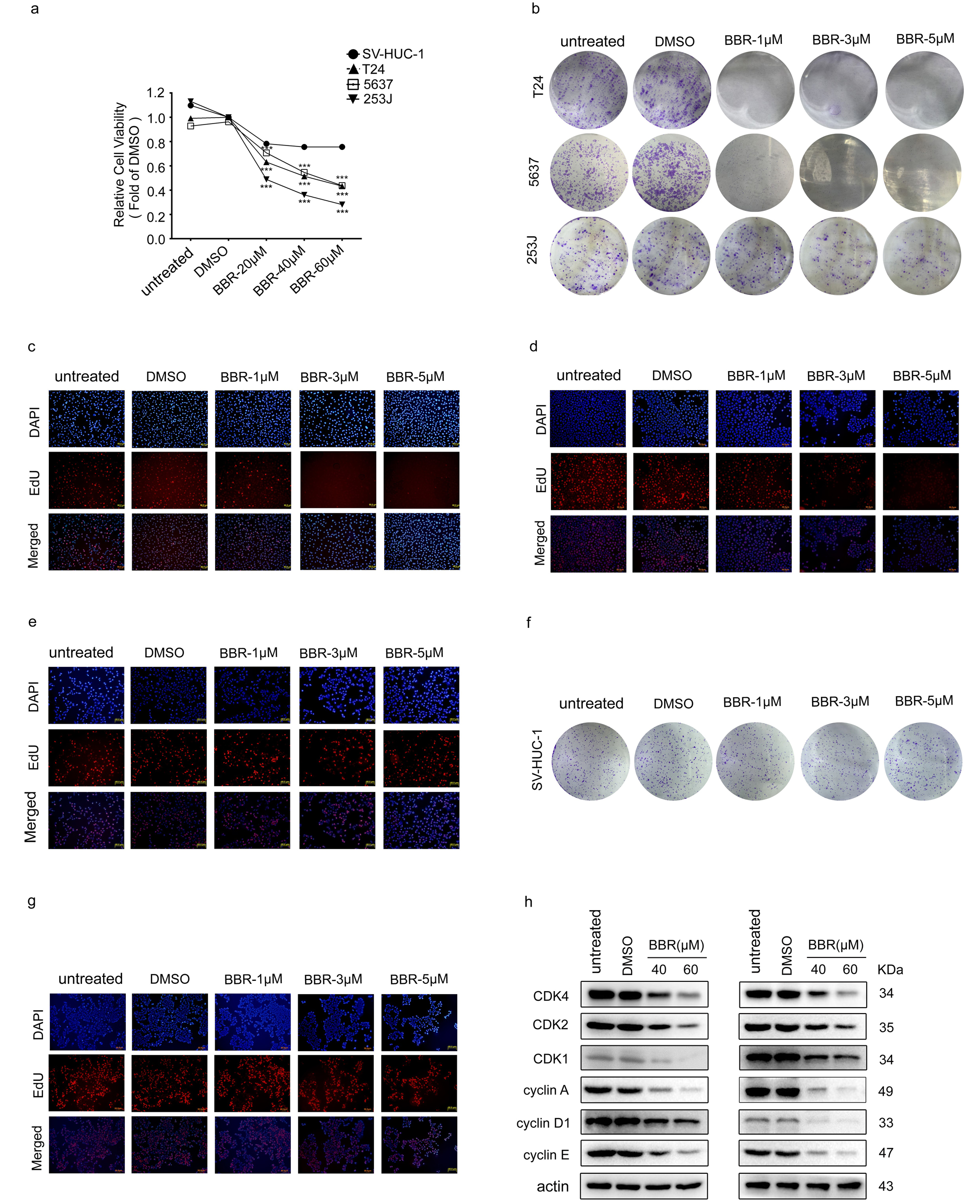
BBR inhibits proliferation and induces cell cycle arrest in BCa cells.
We first evaluated the anti-proliferation effects of BBR on BCa cells. MTT assays showed that BBR significantly decreased cell viability in a dose-dependent manner (Fig. 1a and Supplementary Fig. 1a). The results of colony formation assay also showed that BBR induced a concentration-dependent decrease in the number of cell colonies (Fig. 1b). Moreover, BCa cells treated with BBR exhibited much smaller colonies (Supplementary Fig. 1b). Next, EdU incorporation assays were utilized to detect the effects of BBR on DNA replication. Treatment of BBR significantly decreased the percentage of EdU positive cells in BCa cells (Fig. 1c and Supplementary Fig. 1c-e). Notably, the inhibitive effect of BBR on cell viability, colony formation and DNA replication were much less evident in the human urothelial cells (SV-HUC-1) (Fig. 1a-c and Supplementary Fig. 1a, 1f and 1 g), suggesting that the normal urothelial cells were less sensitive to BBR treatment than that of BCa cells.
Next, we investigated the underlying mechanisms of anti-proliferative effects of BBR in BCa cells. Cell cycle distribution analysis by flow cytometry showed that the effects of BBR on cell cycle varied in BCa cell lines. BBR treatment increased the proportion of cells in G0/G1 phase, accompanied by a decrease in S phase and G2/M in T24 cells (Fig. 1d). While, BBR elevated proportion of S phase in 5637 cells, along with a decline in proportion of G0/G1 and G2/M phase (Fig. 1d). Treatment of 253J cells with BBR resulted in an increase in proportion of G2/M phase (Fig. 1d). Western blots analysis on cell cycle-associated proteins revealed that the protein levels of CDK1, CDK2, CDK4, cyclin A, cyclin D1 and cyclin E were significantly decreased in BBR-treated T24, 5637 and 253J cells (Fig. 1e and Supplementary Fig. 1h).
We then established a xenograft model to further assess the effects of BBR on tumor growth in vivo. Applications of BBR significantly suppressed tumor growth of T24 cells (Fig. 1f). Consistently, the tumor volume and tumor weight were obviously reduced in BBR-treatment group (Fig. 1g). Moreover, immunohistochemistry (IHC) assays showed that BBR administration significantly decreased Ki-67 expression levels (Fig. 1h). Taken together, these results suggest that BBR inhibits the proliferation of BCa cells both in vivo and vitro.
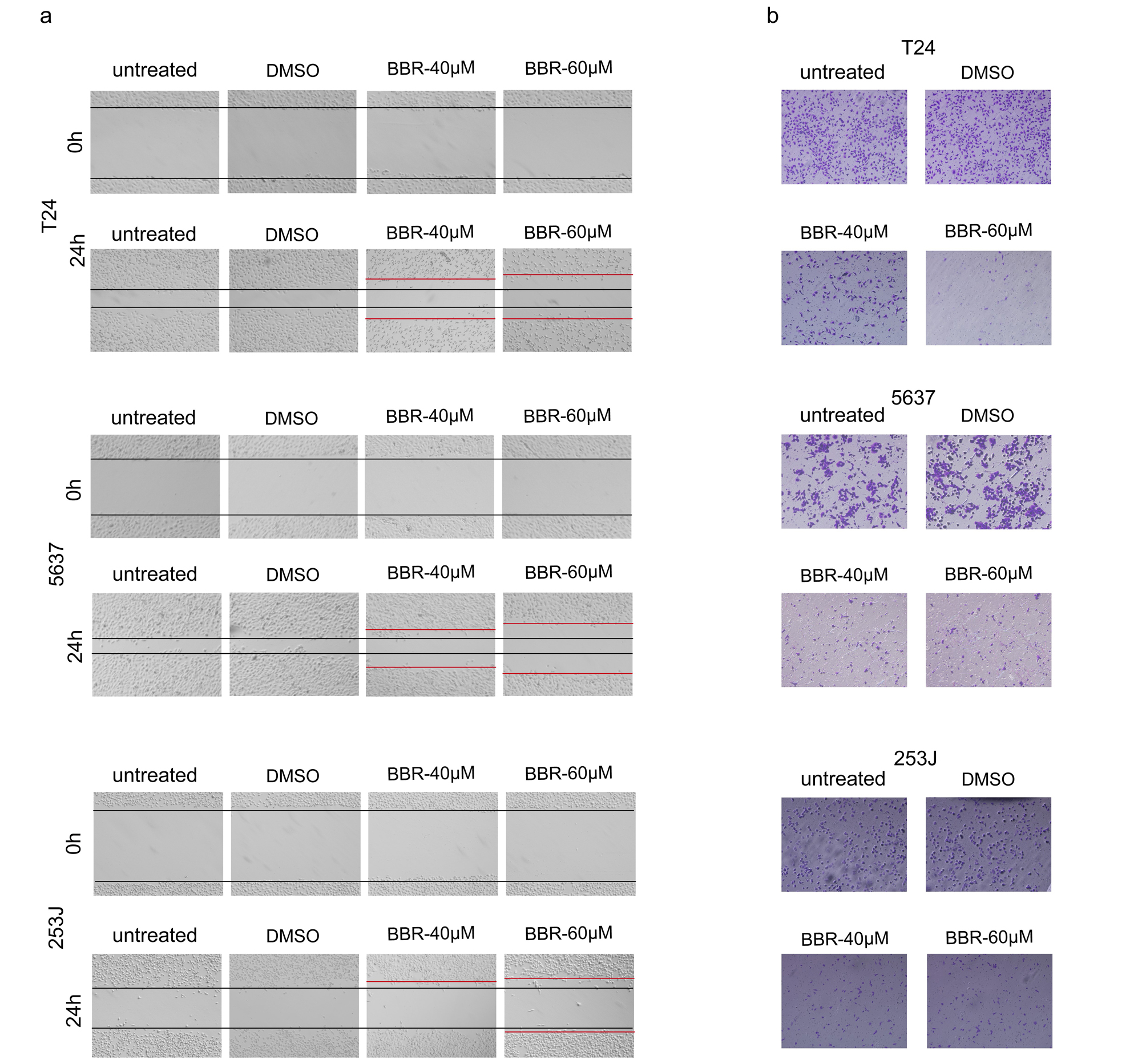
Bbr Suppresses Migration And Invasion Of Bca Cells
We further determined the roles of BRR in migration and invasion of BCa cells. Wound healing assays showed that BCa cells treated with BBR exhibited a lower wound closure rate compared with that of cells treated with DMSO (Fig. 2a and Supplementary Fig. 2a). Transwell assays were further verified the decreased capabilities of migration in BBR-treated BCa cells (Fig. 2b and Supplementary Fig. 2b). Matrigel invasion assays also showed BBR treatment resulted in a smaller number of invasive cells (Fig. 2c-e). These results indicated that BBR significantly attenuated the abilities of migration and invasion of BCa cells. Considering the vital role of Epithelial-Mesenchymal Transition (EMT) in regulating cancer cell migration and invasion, we next detected expression levels of several marker proteins involved in EMT. Western blot analysis revealed that BBR significantly down-regulated the expression of Snail, N-Cadherin and Vimentin in BCa cells (Fig. 2f-h), suggesting that BBR suppresses EMT in BCa cells. Besides, BBR treatment suppressed mRNA expression levels of MMP2 and MMP9 (Fig. 2i), two important genes for tumor metastasis.
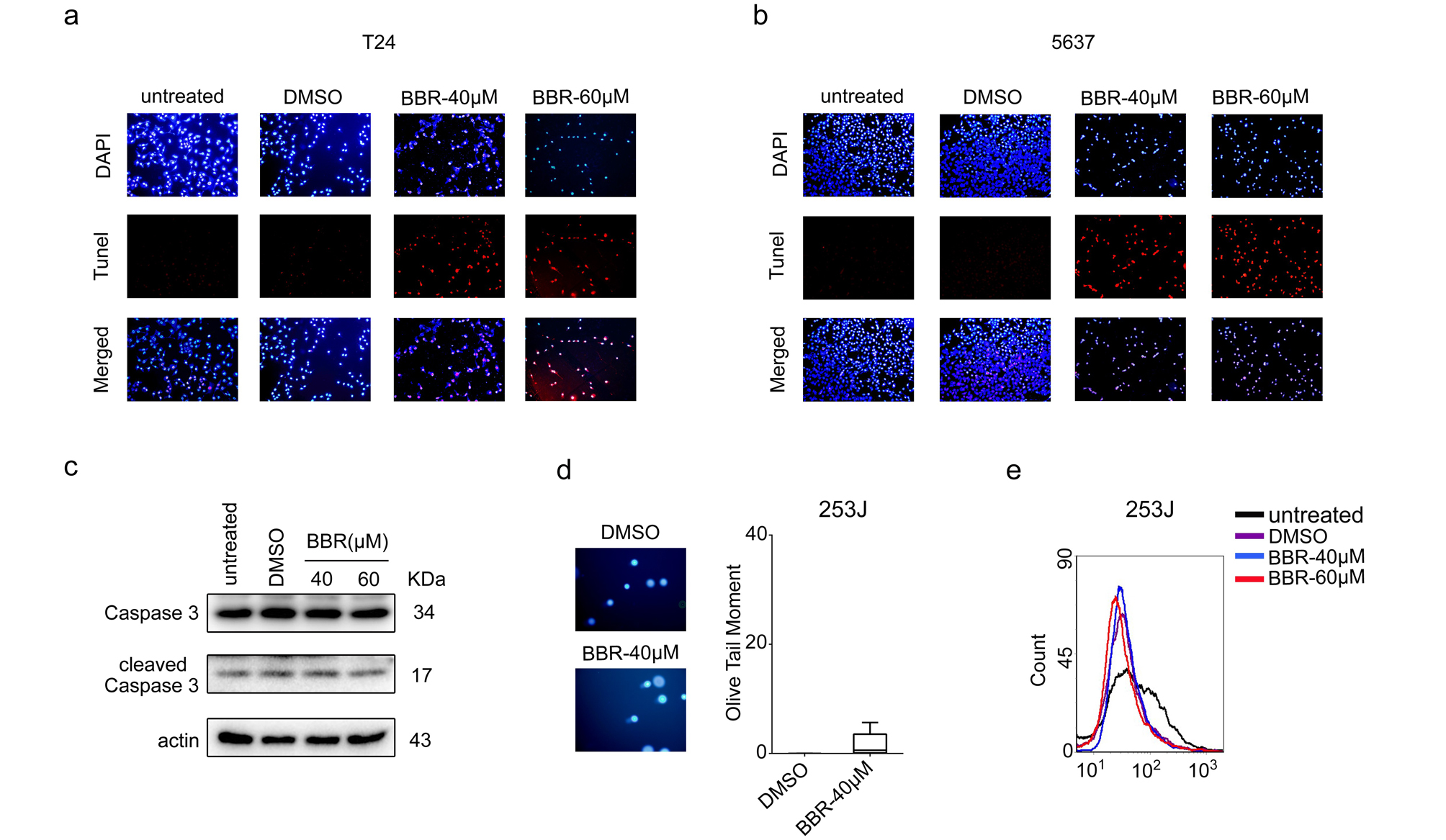
BBR induces apoptosis or senescence in BCa cells in a p53-dependent way
Previous studies reported that BBR exerted anti-proliferative effects through apoptosis induction in various cancer cells. Thus, we next investigated whether BBR caused apoptosis in BCa cells. As shown in Fig. 3a-c, treatment of BBR significantly increased the percentage of apoptotic cells in T24 and 5637 cells, however, no significant changes in apoptotic cells percentage were observed in 253J cells. Consistently, the percentages of Tunel positive cells were significantly increased in BBR-treated T24 and 5637 cells (Fig. 3d and Supplementary Fig. 3a-b), but not in 253J cells (data not shown). Moreover, the levels of cleaved caspase-3, a marker of apoptosis, were up-regulated in T24 and 5637 cells with BBR treatment (Fig. 3e). In contrast, BBR treatment didn’t affect the levels of cleaved caspase-3 in 253J cells (Supplementary Fig. 3c).
BBR was reported to induce cell apoptosis through accumulation of reactive oxygen species (ROS) and oxidative DNA damages. The comet assays showed that the mean olive tail moment values were significantly increased in T24 and 5637 cells treated with BBR (Fig. 3f-g). Moreover, the intracellular ROS levels were also drastically increased with increasing concentrations of BBR in these two cell lines (Fig. 3h), suggesting BBR caused ROS accumulation and DNA damage in T24 and 5637 cells. In contrast, BBR-treated 253J cells displayed similar olive tail moment values and ROS levels with that of DMSO-treated cells respectively (Supplementary Fig. 3d-e), indicating that the underlying mechanisms about proliferative inhibition of BBR might be different between 253J and the other two BCa cell lines. To investigate the mechanisms of BBR in cell proliferation regulation in 253J cells, we next performed SA-β-gal activity assays to evaluate the effect of BBR on cell senescence. The results showed that BBR treatment significantly increased the number of SA-β-gal positive cells (Fig. 3i), substantiating that BBR induces cell senescence of 253J cells.
p53 is considered as one of the most critical tumor suppressors, and previous studies showed that cancer cells harboring wild-type or mutant-type p53 displayed different outcomes of drug-induced cell death24. The results that BBR induced apoptosis in p53 mutant T24 and 5637 cells, while induced cell senescence in p53 wild-type 253J cells prompted us to investigate whether the apoptosis induced by BBR in BCa cells was dependent on p53 status. 253J cell line with stable p53 knock-out (253J p53-KO) was established (Fig. 3j-k). As shown in Fig. 3l, BBR treatment in 253J p53-KO cells caused a significant increase in apoptotic cells percentage compared with that of negative control (253J NC) cells. Besides, knockdown of p53 significantly attenuated the cell senescence caused by BBR in 253J cells (Fig. 3m), suggesting that the cell fate decisions, by BBR, depend on p53 function in BCa cells.
BBR down-regulates the expression and activity of STAT3 in BCa cells.
To further investigate the mechanism that BBR inhibits proliferation of BCa cells, the levels of phosphorylated ERK (p-ERK), phosphorylated AKT (p-AKT) and phosphorylated STAT3 (p-STAT3), which were known as important regulators in cancer progression, were examined. As shown in Fig. 4a, no significant changes in protein levels of p-ERK and p-AKT were observed in BBR treated T24 and 5637 cells. In contrast, both the p-STAT3 and total STAT3 protein levels were significantly decreased in BBR treated T24 and 5637 cells (Fig. 4b). Given that nuclear translocation of activated STAT3 was necessary for transcription of its target genes, the levels of cytoplasmic and nuclear STAT3 and p-STAT3 were detected. As shown in Fig. 4c, both cytoplasmic and nuclear STAT3 and p-STAT3 protein levels were down-regulated by BBR. Furthermore, luciferase analysis confirmed an obvious decrease in transcriptional activity of STAT3 in BBR-treated cells (Fig. 4d). Together, these results suggest that the activity of STAT3 was suppressed by BBR in BCa cells.
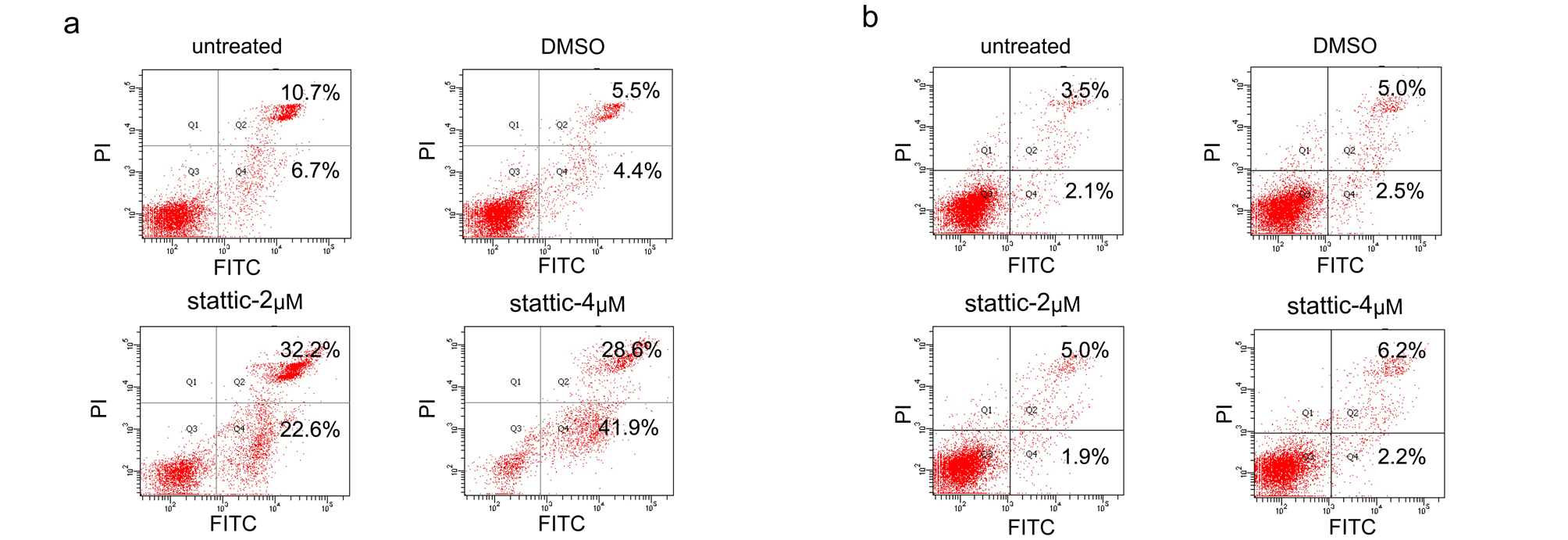
We next sought to evaluate whether STAT3 inhibition was responsible for BBR-mediated proliferation suppression in BCa cells. Stattic, a STAT3 inhibitor, significantly inhibited cell viability in a concentration-dependent manner (Fig. 4e). Importantly, similar with that of BBR, stattic treatment induced apoptosis in T24 cells, while induced cell senescence in 253J cells (Fig. 4f-4 g and Supplementary Fig. 4a-b). Moreover, stattic treatment in 253J p53-KO cells also significantly induced apoptosis accompanied by reduction of cell senescence (Fig. 4h-4i), resembling that of BBR treated cells. Altogether, these results suggest that the proliferation inhibition, apoptosis and senescence induction caused by BBR in BCa cells are potentially dependent on downregulation of STAT3.
Bbr Inhibits Stat3 Activation Through Suppressing Jak1
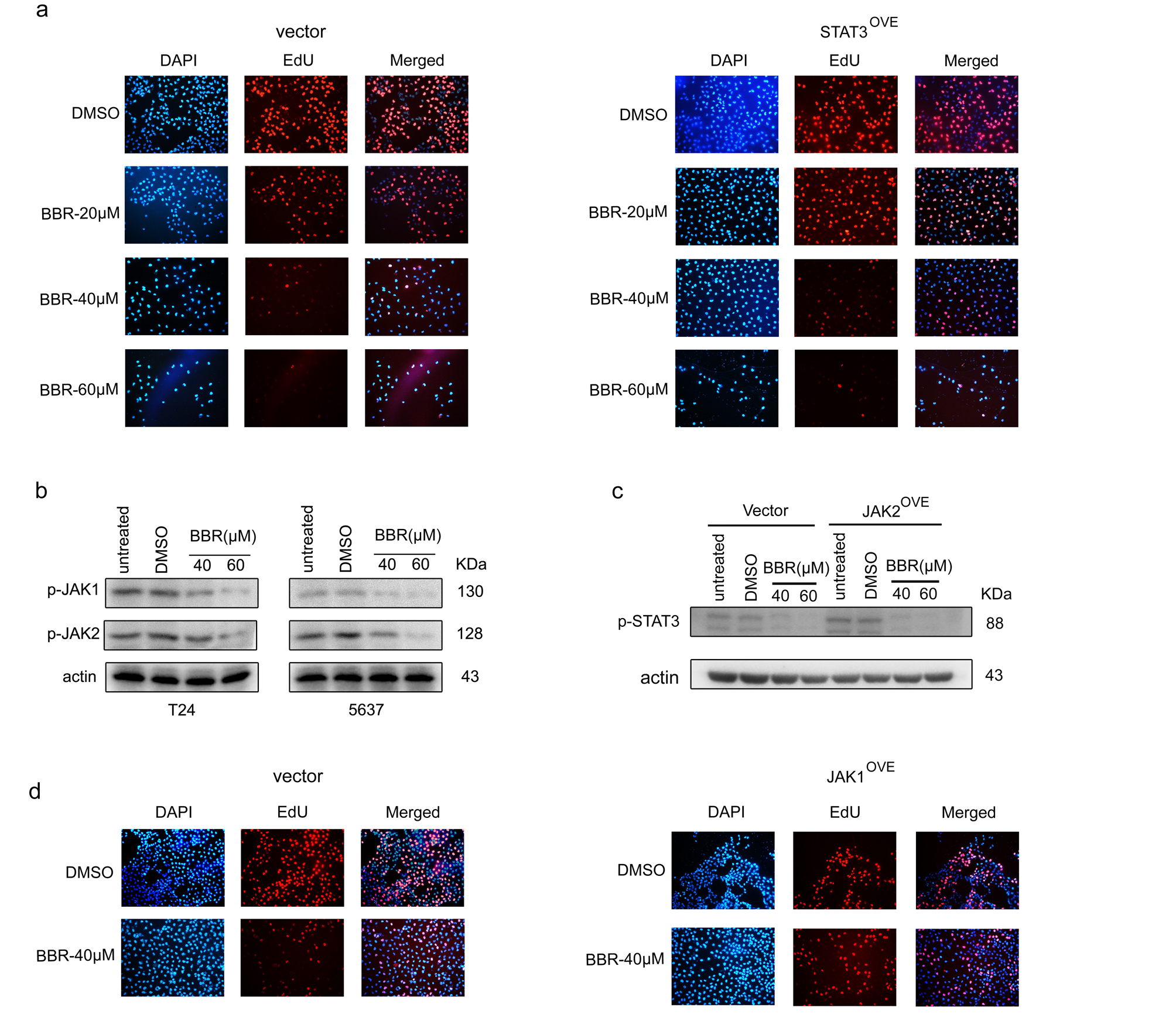
To further investigate whether the anti-tumor effect of BBR on BCa cells was mediated by STAT3 expression inhibition, we established STAT3 overexpressed T24 and 5637 cells (STAT3OVE) (Fig. 5a). Western blot analysis showed that overexpression of exogenous STAT3 significantly restored the reduced endogenous total STAT3 protein level caused by BBR (Fig. 5b). Of note, BBR also significantly decreased the exogenous STAT3 expression (Fig. 5b). MTT assays showed that expression of exogenous wild-type STAT3 didn’t rescue BCa cells from proliferative inhibition caused by BBR (Fig. 5c). EdU incorporation assays showed that the number of EdU positive cells was statistically increased in STAT3OVE cells than that of control cells under BBR treatment, however, it still significantly less than that of DMSO treated cells (Fig. 5d and Supplementary Fig. 5a), indicating the protective effect of introduction of exogenous STAT3 on DNA replication is not significantly effective to attenuate the efficacy of BBR in BCa cells. Consistently, overexpression of STAT3 didn’t obviously restore p-STAT3 levels downregulated by BBR (Fig. 5e). Together, these results indicate that BBR-induced proliferation and STAT3 activity inhibition are not only mediated by decreasing STAT3 expression.
Given the critical role of Janus kinases (JAKs) in activating STAT3, we next examined the expression levels of JAK1 and JAK2. As shown in Fig. 5f and Supplementary Fig. 5b, BBR significantly decreased both the total and phosphorylated protein levels of JAK1 and JAK2 in T24 and 5637 cells. Importantly, overexpression of JAK1, but not JAK2, significantly restored p-STAT3 levels downregulated by BBR (Fig. 5g-i and Supplementary Fig. 5c), indicating that JAK1 is a key target through which BBR exerted anti-tumor effects on BCa cells. As expected, overexpression of JAK1 significantly attenuated the BBR-induced proliferation inhibition, cell cycle arrest and apoptosis (Fig. 5j-m and Supplementary Fig. 5d). Together, these results suggested that, BBR suppresses JAK1 expression to inhibit STAT3 activation in BCa cells.
Bbr Suppresses Jak1-stat3 Signaling Via Upregulation Of Mir-17-5p
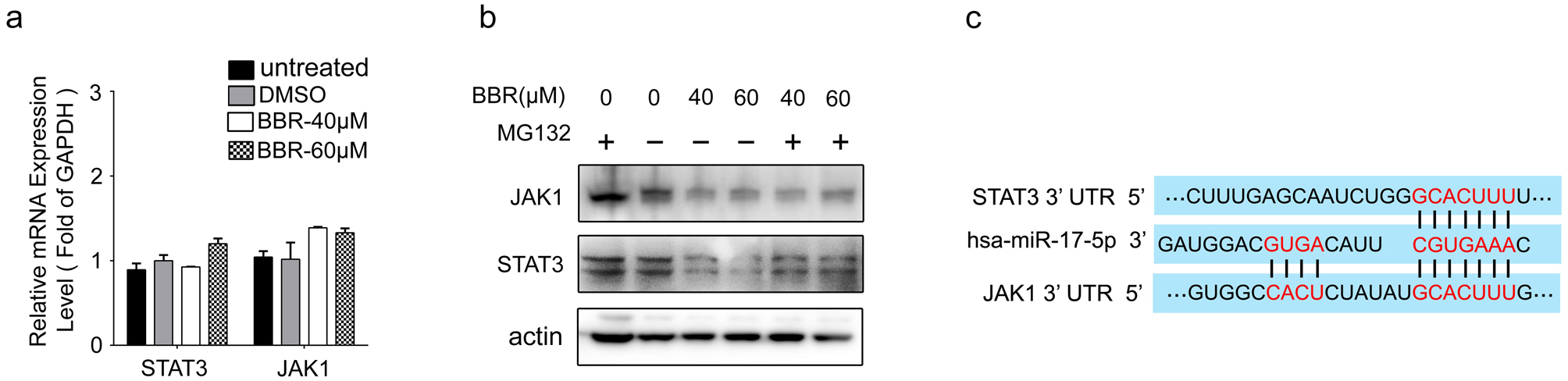
To elucidate the mechanisms that BBR downregulates JAK1 and STAT3 expression, we first detected their mRNA levels. As shown in Fig. 6a and Supplementary Fig. 6a, BBR treatment did not decrease mRNA levels of JAK1 and STAT3 in 5637 and T24 cells. Moreover, the decreased protein levels of JAK1 and STAT3 caused by BBR treatment could not be restored by MG132 (Supplementary Fig. 6b). All these results suggest that downregulation of JAK1 and STAT3 expression induced by BBR was not mediated by regulating transcription or protein degradation.
MicroRNAs (miRNAs) degrades or inhibits translation of downstream target genes via binding to complementary mRNAs. Accumulating evidence has proved that dysregulated miRNAs induced by drug treatment were important regulators of cell fate in cancer progression25. Thus, we then investigated whether BBR downregulated JAK1 and STAT3 via miRNAs. The online softwares– TargetScan and miR WALK were utilized to predict miRNAs targeting both JAK1 and STAT3, and 5 candidate miRNAs were identified (Fig. 6b). qRT-PCR assays showed that only miR-17-5p level was upregulated by BBR treatment (Fig. 6c). Further analysis revealed that BBR promoted expression of miR-17-5p in a concentration-dependent manner (Fig. 6d). Importantly, transfection of miR-17-5p mimics significantly decreased protein levels of JAK1 and STAT3 in T24 and 5637 cells, leading to downregulate p-STAT3 expression (Fig. 6e). Moreover, miR-17-5p mimics significantly inhibited cell proliferation (Fig. 6f) in 5637 cells, similar with that of BBR. To determine whether miR-17-5p directly binds to 3’ UTR regions of JAK1 and STAT3 as in-silico prediction (Supplementary Fig. 6c), dual-reporter luciferase assays were performed. As shown in Fig. 6g, the activities of reporters containing wild-type JAK1 and STAT3 3’UTR, but not the miR-17-5p binding sites mutant 3’UTR, were decreased by transfection of miR-17-5p mimics. Collectively, these data indicate that miR-17-5p directly binds to 3’ UTR of JAK1 and STAT3 and inhibits their expression in BCa cells.
We then investigated whether BBR downregulated JAK1 and STAT3 expression via miR-17-5p regulation. As shown in Fig. 6h, transfection with miR-17-5p inhibitors partially blocked the decreased expressions of JAK1, p-STAT3 and STAT3 caused by BBR treatment. Furthermore, luciferase reporter assays showed that the wild-type JAK1 and STAT3 3’UTR activities were significantly decreased by BBR. However, these inhibitory effects were abolished by mutating the miR-17-5p binding sites (Fig. 6i), suggesting that BBR perturbed JAK1-STAT3 pathway via upregulation of miR-17-5p.
The effect of BBR on miR-17-5p-JAK1-STAT3 pathway in vivo.
Last, we examined JAK1 and STAT3 expression levels in xenograft tumors. The results showed that mRNA levels of JAK1 and STAT3 were not significantly altered by BBR treatment (Fig. 7a-b), however, miR-17-5p expression was obviously upregulated in BBR-treatment tumors (Fig. 7c). Moreover, the results of western blot and IHC analyses on xenograft tumors confirmed that BBR decreased JAK1, STAT3 and p-STAT3 protein levels in vivo (Fig. 7d-e). Collectively, these results suggest that BBR promotes miR-17-5p expression to suppress JAK1 and STAT3 both in vivo and vitro.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}