Bacterial strains, culture media and cultivation conditions
Bacterial strains and cultivation conditions, used in this study, are listed in Table 3.
S-layer-carrying Lb. brevis SF9B was previously characterised by Banić et al. (2018). Strains are deposited in the Culture Collection of the Laboratory of Antibiotic, Enzyme, Probiotic and Starter Culture Technologies, Faculty of Food Technology and Biotechnology, University of Zagreb (CIM-FFTB) and are maintained as frozen stocks at -80 °C in appropriate medium supplemented with 15% (v/v) glycerol.
Human cell line, culture medium and cultivation conditions
Enterocyte-like Caco-2 cells were donated by the Ruđer Bošković Institute, Zagreb, Croatia. Caco-2 cells were grown as monolayer cultures in RPMI 1640 medium (GIBCO, USA), supplemented with 15% of the fetal bovine serum (GIBCO, USA) and 4500 mg/l of glucose. Cells were grown up to confluence at 37 °C and 5% of CO2 in T-flasks, trypsinised and seeded into 24-multiwell plates. Prior to experiments, cells reached sub-confluence.
DNA isolation and PCR analysis
Total genomic DNA, both for PCR analysis of the bacteriocin genes or WGS, was extracted according to the method of Leenhouts et al. (1990) with minor modifications. The purity and concentration of the extracted DNA were then determined by using a BioSpec-Nano spectrophotometer (Shimadzu, Kyoto, Japan) and the extracted DNA was stored at -20 °C. PCR screening for the prevalence of bacteriocin structural genes was performed with primers listed in Ben Omar et al. (2008). Therefore, amplification of DNA fragments was performed in 50 µl reaction mixtures containing 25 µl of Emerald Amp MAX HS PCR Mastermix Premix (TaKaRa, Ohtsu, Japan), 200 nmol/l of each oligonucleotide primer, 300 ng of DNA template and EmeraldAmp dH20. A negative control, which contained all reagents except the DNA template, was used to detect contamination or non-specific amplification. The amplification was carried out in an Eppendorf Mastercycler personal thermal cycler (Eppendorf, Germany) using the conditions described by the Ben Omar et al. (2008). PCR-amplified products were separated by electrophoresis in a 1% agarose gel, stained with ethidium bromide (0.5 μg/ml) and visualised on a MiniBIS Pro transilluminator (DNR Bio-Imaging Systems Ltd., Jerusalem, Israel) at 254 nm and images were captured by the GelCapture software version 7.1 (DNR Bio-Imaging Systems Ltd., Jerusalem, Israel).
Whole genome sequencing and identification of genesencodingbacteriocins
Genomic DNA was prepared according to Frece et al. (2009). Genome sequencing was done using a paired-end approach as essentially described in Banić et al. (2018). Briefly, the Nextera DNA Library Preparation Kit (Illumina, San Diego, CA, USA) was used to construct a library. The library was processed with the Illumina cBot and sequenced on the MiSeq2500 (Illumina, San Diego, CA) pair-end with 300 cycles per read. Contigs were classified as belonging to Lb. plantarum when obtaining the best blastn v2.2.27 hit (Altschul et al., 1990) in the NCBI nt database. RAST server, which identifies protein-encoding, rRNA and tRNA genes, assigns functions to the genes, and predicts which subsystems are represented in the genome (Aziz et al., 2008), was used for the annotation, and the distribution and categorization of all sequenced genes. The assembled contigs were compared with so far identified bacteriocins in the NCBI using the tblastn v2.2.27. To further supplement the annotation, BAGEL4 sofware was used to predict genes related to bacteriocin synthesis (van Heel et al., 2018). The input file was the genome sequence of Lb. plantarum SF9C in a fasta file. Conserved genes associated with the bacteriocin synthesis were retrieved using the RAST server (Aziz et al., 2008). Additionally, WGS were pairwise aligned with “run-mummer3” to detect alignments and SNPs. The plot was computed with R package hclust, based on SNP frequency. Further, the 3D structure homology modelling was done using the SWISS-MODEL server (https://swissmodel.expasy.org/) based on the alignment of the amino acid sequences of the core peptides, generated from BAGEL4 software. Additionally, helix properties of each two plantaricin were calculated using heliQuest web server (Gautier et al., 2008).
In vitro assays
Testing of antimicrobial activity
The antimicrobial activity of the overnight grown culture of Lb. plantarum SF9C and Lb. brevis SF9B strains against L. monocytogenes ATCC®19111™ and S. aureus 3048 was tested by agar spot test and well-diffusion method. The agar spot test was performed according to Leboš Pavunc et al. (2013). The ratio of the inhibition diameter (ID) to the spot culture diameter (CD) was calculated to determine the effective inhibition ratio (EIR) of SF9C strain: ((ID-CD)/CD). Furthermore, the agar well-diffusion method, previously described by Kos et al. (2008) was applied for the analysis of antimicrobial activity of the cell free supernatant (CFS). CFS was recovered by centrifugation, filtered through a 0.22 µm sterile filter (Millipore Corporation, Billerica, MA, USA) and concentrated up to 5-fold in an Amicon cell concentrator (Amicon, Beverly, MA, USA) equipped with a selective (10 kDa) membrane. The proteinaceous nature of potential inhibitory compounds in CFS was examined by treatment with Proteinase K (Invitrogen, Carlsbad, CA, USA) at a concentration of 1 mg/ml during 2 h at 37 oC and by heating the samples at 100 ˚C/30 min, according to Elayaraja et al. (2014). Statistical analysis was carried out using ANOVA and the results are reported as mean values of three individual experiments ± standard deviation. One-way analysis of variance (ANOVA) and Tukey tests were performed using VassarStats software to determine significant group differences and means were considered as statistically significant if P < 0.05.
Evaluation of the antibacterial activity after cocultivation with the targeted pathogens
The influence of cocultivation of Lb. plantarum SF9C with L. monocytogenes ATCC®19111™ and S. aureus 3048 on bacteriocin activity of SF9C strain was performed according to Kos et al. (2008) with slight modifications. The number of viable cells was determined by spot-plate method using the corresponding selective media for each strain: MRS for lactobacilli; Baird-Parker (Oxoid, Hampshire, UK) for S. aureus and ChromoBio (Biolab Diagnostic Laboratory, Budapest, Hungary) for L. monocytogenes, in 2 hour intervals during the first 10 hours, and after 22, 24 and 48 h of the incubation. Plates were incubated for 24 hours at 37 °C and the number of viable cells was expressed as log CFU/ml. Also, during the experiment, the antibacterial activity of SF9C strain, in monoculture and coculture, was tested by agar spot test as described above. Experiments were conducted in triplicate and values were expressed as the mean ± standard deviation. One-way analysis of variance (ANOVA) and Tukey tests were performed for statistical analysis.
Pathogen competition and exclusion assay by Lb. brevis SF9B and Lb. plantarum SF9C on Caco-2 cell line
For exclusion and competition assay experiments, Caco-2 cells were routinely grown in 24-well culture plates until confluent differentiated monolayers were obtained. Cellular monolayers were carefully rinsed three times with PBS (pH 7.4) before addition of the bacterial cells. Two separate protocols were followed to assess the ability of viable lactobacilli strains to inhibit E. coli 3014, S. Typhimurium FP1 adhesion to Caco-2 cells. For both assays, Lactobacillus strains and pathogens were routinely cultivated; the cells were harvested and prepared in PBS (pH 7.4) to reach A620 = 1 (approximately 1 x 109 CFU/ml). The competition assay was performed according to the procedure described by Uroić et al. (2016) with few modifications. Lactobacilli and pathogens were co-incubated with Caco-2 monolayer for 1 h. For exclusion assays, Lactobacillus strains were cultured with Caco-2 monolayer for 1 h. Following 1 h incubation, Caco-2 monolayers were gently washed three times with PBS (pH 7.4); pathogens were added and incubated for another 1 h. A 1.0 ml aliquots of the monospecies cultures of pathogenic bacteria together with 1.0 ml of EMEM per well were used as the controls in both assays. In all the above treatments, post-incubation removal of the non-adhered bacterial cells was executed by removing the bacterial suspension and washing the Caco-2 monolayers three times with PBS (pH 7.4). The Caco-2 cells were then lysed by addition of 0.25% (v/v) Triton X-100 (AppliChem, Darmstadt, Germany) solution at 37 °C for 10 min in order to collect the adherent bacterial cells, and the total numbers of viable adhering Lactobacillus, E. coli and S. Typhimurium were determined by spot-plate method on MRS, Rapid (Biorad, Dubai, United Arab Emirates) and XLD (Biolife, Milano, Italy) agar plates, respectively. The efficiency of pathogen exclusion of Lactobacillus strains was assayed in three biologically independent experiments each with three replicates.
In vivo animal trial
Preparation of Lb. brevis SF9B and Lb. plantarum SF9C strains and administration to rats
Bacterial cultures Lb. brevis SF9B and Lb. plantarum SF9C were grown in 5 ml of MRS broth at 37 °C under anaerobic conditions until the OD value reached 1.0 at 620 nm. The as-prepared cultures were mixed in 1:1 (v/v) ratio and inoculated (4%) in 50 ml of MRS broth. After overnight incubation at optimal conditions, the cells were harvested by centrifugation at 5000 g for 10 min, suspended in saline solution and the presence of both strains was microscopically examined. The bacteria suspensions were prepared daily to ensure viability and the CFU was controlled to maintain strictly the number of CFU administered by a rat as it is described in the next chapter.
Experimental animals
Three-months-old male highly inbred Y59 strain rats, weighing 200 to 250 g, (http://www.informatics.jax.org/external/festing/rat/docs/Y59.shtml), obtained from our breeding within the Department of Animal Physiology, Faculty of Science, University of Zagreb, were used in this study. The animals were maintained under a 12/12-h light-dark cycle with free access to food and water and standard housing conditions (room temperature around 25 °C and 60% humidity). They were fed a standard laboratory diet (4 RF 21, Mucedola, Settimo Milanese, Italy) and tap water ad libitum. Maintenance and care of all experimental animals were carried out according to the guidelines in force in the Republic of Croatia (Law on the Welfare of Animals, NN135/06 and NN37/13) and in accordance with EU Directive 2010/63/EU for animal experiments (OJEU, 2010) and carried out in compliance with the Guide for the Care and Use of Laboratory Animals, DHHS Publ. # (NIH) 86-123. The experimental procedure was approved by the Bioethics Committee of the Faculty of Science, University of Zagreb, Croatia (No. HR-POK-012).
Rat study design and sample collection
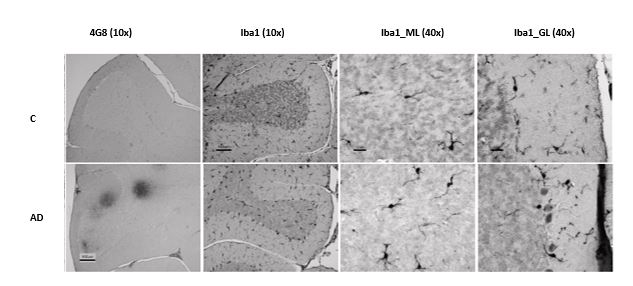
Male rats belonging to the Y59 inbred strain were randomly divided into 2 equally sized trial groups and housed three per cage in stainless-steel cages, under the same controlled conditions. The rats were treated daily for five consecutive days with a single dose (3x109 CFU/ml) of Lb. brevis SF9B and Lb. plantarum SF9C strains suspended in saline solution, starting 24 h after the last treatment as follows: (a) first trial group represented a model of induced aluminium toxicity which was established by intraperitoneally injecting AlCl3 (10 mg/kg) and D-galactose (60 mg/kg) as described by Ulusoy et al. (2015) and (b) second group served as healthy (control) group and was injected comparatively with saline solution in the same manner. No side effects were reported following Lactobacillus administration. In order to evaluate the AchE activity, which requires a brain sample, rats had to be sacrificed. Before the sacrifice rats were anesthetized using a mixture of ketamine (Narketan®10, Vetoquinol AG, Belp Bern, Switzerland) at dose of 75 mg/kg with xylazine (Xylapana® Vetoquinol Biowet Sp., Gorzow, R. Poland) at dose of 10 mg/kg. The intestinal mucosal content from each sacrificed rat was scraped and specimens were kept frozen at -80 °C until the analysis. The brain was removed and frozen at -80 °C or kept in buffered formaldehyde until the analysis. The brain tissue homogenates were used to assess acetylcholinesterase (AChE) activity by colorimetric method. AchE activity is expressed in mol/min/g tissue. The brain samples are prepared according to standard paraffin procedure. Changes related to early-stage Alzheimer’s disease were also (un)confirmed by immunohistochemistry used primary antibodies Purified-β-Amyloid, 17-24 Antibody (4G8) diluted 1:2000 (BioLegend, San Diego, CA), Phospho-PHF-Tau (pSer202 + Thr205) Monoclonal Antibody (AT8) diluted 1:500 (Thermo Fisher Scientific, Waltham, MA, USA) and Iba1 diluted 1:250 (Wako Pure Chemical Industries, Japan). Photomicrographs were recorded using a digital camera (AxioCam ERc5s, Zeiss) and processed by a computer program morphometric image analysis (AxioCam ERc5s-ZEN2). The faecal samples were collected from the cages before starting the treatment and on the 3rd and 10th day following the last Lactobacillus administration in triplicates. Faecal samples were stored at -80 °C until analysis as described in the next chapter.
Bacterial 16S rRNA sequencing and processing using QIIME
Faecal samples were collected from rats at the end of the study and used to purify the total genomic DNA using a commercial DNA extraction kit Maxwell DNA Tissue Kit with automated extraction platform, Maxwell® 16 Research System instrument (Promega, USA). The final equimolar pool was sequenced on the Illumina MiSeq platform. PCR reactions and 16S sequencing were performed at the Molecular Research LP (MRDNA, Shallowater, Texas USA). The MiSeq instrument (Illumina) was used for sequencing the 16S amplicons following the manufacturer’s instructions at MRDNA described by Garcia-Mazcorro et al. (2018) with slight modifications. Raw 16S data were obtained from Illumina’s basespace as FASTQ files and analysed using the QIIME 2 pipeline using the procedure as described in the moving pictures tutorial (https://docs.qiime2.org/2018.11/tutorials/moving-pictures/).
PCR –DGGE analysis
PCR-DGGE analysis was performed according to Leboš Pavunc et al. (2012) with slight modifications in order to check the presence of the Lb. plantarum SF9B and Lb. brevis SF9C in faeces of Lactobacillus fed rats. DNA was extracted directly from faecal samples of healthy rats for culture-independent PCR-DGGE analysis, as well as from the bacterial colonies, isolated on MRS agar plates for culture-dependent PCR-DGGE analysis, from faeces of healthy rats sampled before feeding (control), and 3rd and 10th day after application of Lactobacillus SF9B and SF9C strains. In both cases, DNA was isolated using Maxwell DNA Cell Kit with automated extraction platform, Maxwell® 16 Research System instrument (Promega, USA). The V2-V3 region of the 16S ribosomal DNA gene of bacteria in the faeces contents or from pure cultures of lactobacilli was amplified with primers HDA1-GC and HDA2. To identify the lactobacilli, recovered from rat faeces, the V2-V3 region of the 16S rRNA gene of the strains was amplified. The amplicons were sequenced using ABI PRISM® 3100-Avant Genetic Analyzer (Applied Biosystems). A search of sequences deposited in the GenBank DNA database was conducted by using the BLAST algorithm. The identities of the isolates were determined based on the highest score.
Statistical analysis
All the experiments were repeated three times and the results were expressed as means of three independent trials ± standard deviation (SD). Statistical significance was appraised by one-way analysis of variance. Pairwise differences between the means of groups were determined by the Tukey HSD test for post-analysis of variance pairwise comparisons (http://vassarstats.net/test). Statistical differences between groups were considered significant when P values were less than 0.05.
{kind=link}
{kind=link}