Reagents and animals
Hyaluronic acids (HA, 1.3 MDa and 2.3 MDa) were purchased from Novozymes (Beijing, China). Dopamine hydrochloride (DA) was acquired from Tansoole Co., Ltd. (Shanghai, China). 2-Amino-2-(hydroxymethyl)-1, 3-propanediol (Tris-HCl, pH 8.5) was purchased from Beyotime (Shanghai, China). Streptomycin, penicillin and 0.25 wt.% trypsin with 0.02 wt.% Ethylene diamine tetraacetic acid (EDTA) were purchased from Gibco BRL (Gaithersburg, MD). High glucose DMEM medium (1×) were purchased from Zhejiang Senrui Co., Ltd. (Huzhou, China).
Female SD rats ranging from 220 g to 250 g used in the SCI model were purchased from SLAC Laboratory Animal Co. Ltd. (Shanghai, China). All animal experiments and procedures were approved and in compliance with the Institutional Animal Care and Use Committee at Zhejiang University.
Fabrication of hydrogel
Basic hydrogel materials were prepared according to previous articles. Briefly, to introduce -CHO at -OH sites of chains, HA (2.3 MDa, 500 mg, 1.25 mmol) was dissolved in ultrapure water (150 mL) and lucifugally oxidized by NaIO4 (10 mg·mL− 1) with a mole ratio of 1:0.5 (NaIO4:HA). At the end of 2 continuous days of stirring, the reaction system was stopped by adding ethylene glycol (600 µL) for 1 h. After dialysis, the product was lyophilized and -CHO modified HA (HA-CHO) was obtained. To introduce -NH2 into HA, adipic dihydrazide (ADH) was utilized as the source. HA (1.3 MDa, 270 mg, 0.675 mmol) and ADH (20.25 mmol, 4.64 g) at a mole ratio of 1:30 (HA:ADH) was dissolved in ultrapure water (150 mL). With an adjusted pH of 6.8, 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC, 0.96 g, 5 mmol) and 1-hydroxybenzotriazole (HOBt, 0.675 g, 5 mmol) in dimethyl sulfoxide-water (1:1) were added into the reaction system separately. The pH at 6.8 was maintained for the following 4 h and changed to pH 7.0 to end up the reaction. The HA-ADH product was then lyophilized and collected.
Superficial modification of hydrogel
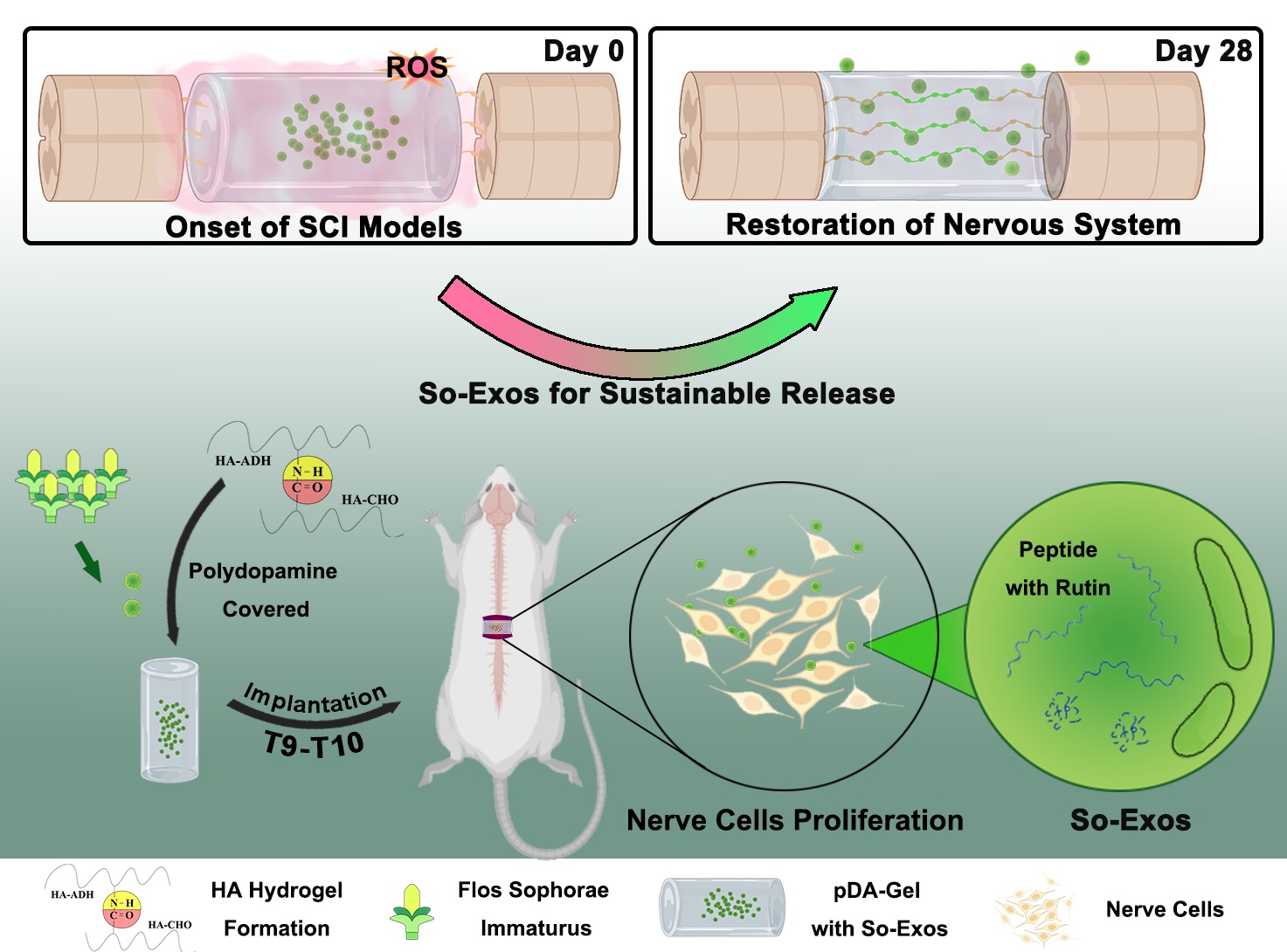
HA-CHO and HA-ADH were dissolved in phosphate buffered saline (PBS) at 20 mg·mL− 1 and 15 mg·mL− 1, respectively. Basic hydrogels were formed with the mixed solution at an equal volume of 30 µL. After placing at 4 ℃ overnight, hydrogel was lyophilized and collected. To further modify the hydrogel, lyophilized hydrogel was swelled in PBS and sterilized under the ultraviolet light overnight. Swelled basic hydrogel were soaked in the polydopamine hydrochloride (pDA) solution at 1 mg·mL− 1 in Tris buffer (pH 8.5). The superficial modification should take place on an orbital shaker overnight. The pDA-Gel could be obtained through lyophilization.
Characterization of basic hydrogel materials and the modified materials
The lyophilized HA-CHO was stirred with HBr and pulverized into powder for Fourier transform infrared spectroscopy (FTIR) detection to prove the accomplishment of oxidation of carbonyl group at the site of -OH in the product. The HA-ADH modification was confirmed via 1H nuclear magnetic resonance (NMR). HA-ADH, HA, and ADH were dissolved in deuterium oxide (D2O). To observe the internal porous structure and superficial modification of the pDA-Gel, scanning electron microscope (SEM) (Nova Nano 450, Thermo FEI, Czech) were used. Both the Gel and the pDA-Gel were sliced and covered with gold in vacuum for examination.
Isolation of Sophora exosomes (so-exo)
All Flos Sophora Immaturus were bought from markets. Most of so-exos were naturally stored in the mesoderm of Flos Sophora Immaturus. To prepare so-exos, appropriate sinking buffer was required to penetrate the outer face and permeate the whole mesoderm of plant so as to swell the plant cell. 4-Morpholine ethanesulfonic acid (MES) buffer was known as preservation of antioxidant factors in plants. After sinking the sophora in MES buffer (pH 6.0) for 24 h, the extracted juice was collected from swelled sophora via centrifugation at 2000 g for 20 min. The rough purification of soak extraction was employed through centrifugation at 3000 g and 10000 g for 30 min each to remove cells and debris. The fine purification of supernatant was employed through centrifugation at 100000 g for 70 min twice and an extra PBS resuspension centrifugation. Pelleted so-exos were resuspended in 300 µL PBS. All purification was under the condition of 4 ℃.
Characterization of so-exos
Structural morphology and size confirmation were affirmed via transmission electron microscopy (TEM). 5 µL of so-exo solution was added on formvar/carbon-coated 200-mesh copper electron microscopy grids. After incubation at room temperature for 3 min, standard uranyl acetate staining was performed to fix the whole structure. Before the TEM observation, grids were semi-dried at room temperature.
Quantification and labeling of so-exos
Rutin components inside so-exos were detected by high performance liquid chromatography (HPLC, Agilent Technologies 1200 series). Rutin was detected by photo-diode array detector at 257 nm. The chromatographic column was C18 column (5 µm, 150 mm×4.6 mm) (Diamonsil, Dikma, China). The mobile phase was methanol: 1% glacial acetic acid solution, 40:60; Flow rate should be 1 mL·min− 1; Temperature was 25 ℃. Micro BCA protein assay kit (Thermo Fisher Scientific, USA) was used to detect exosome concentration. The ratio between rutin dosage, particle number and protein content were calculated through conversion.
Adhesive ability of so-exos
Both Gel and pDA-Gel were fully swelled in PBS and sterilized under ultraviolet overnight. Gels were washed with 75% ethyl alcohol and PBS for further sterilization. So-exos were resuspended in PBS to a concentration of 5 mg·mL− 1. Each gel was injected with 20 µL of so-exo suspension. Incubation of the so-exos encapsulated gel was conducted at 37 ℃ for 1 h and at 4 ℃ overnight to strengthen the adhesiveness of exosomes as far as possible. Thus, so-exos encapsulated hydrogels were fully prepared for adhesive ability analysis and in vivo implantation. As for adhesive ability analysis, former prepared gels were carefully sunk in PBS for 5 min and rinsed with PBS twice the next day. Afterwards, gels were diluted to 250 µL and vibrated in ultrasound. Once more, vibration was employed to ensure the full extraction of encapsulated exosome. Protein amounts were detected via micro BCA. Blank gel group injected with PBS should be set up to eliminate the material disturbance. As for in vivo implantation, gels were prepared 1 day before the SCI model establishment.
Evaluation of cellular viability, intercellular ROS level and adenosine triphosphate (ATP) activity
SH-SY5Y cells were cultured in high glucose DMEM medium containing 10% FBS, 1% penicillin and streptomycin (100×) with 5% CO2 at 37 ℃. To evaluate cellular viability after diverse treatments, cells were seeded in 96-well plates (5000 cells per well). After the attachment of cells on the plate bottom, medium was replaced with fresh FBS-free medium containing so-exos, rutin with a gradient rutin concentration (0.5, 0.25, 0.125 mg·mL− 1). After 24 h incubation under hypoxia condition (1% O2, 5% CO2, 37 ℃), the plate was rinsed with FBS-free medium. The cell counting kit-8 (cck8) was used to evaluate the cell viability and the detection was manipulated at 450 nm.
The intercellular ROS level was tested using 2,7-dichlorodi-hydrofluorescein diacetate (DCFH-DA) as the ROS detector. After incubation and rinse, cells were incubated with 1:1000 DCFH-DA in FBS-free medium at 37 ℃ for 20 min. The detection was manipulated on SpectraMax M5 (Molecular Devices, USA) after rinsing. The excitation and emission wavelengthes were 488 and 525 nm respectively. The ATP activity evaluation was tested via ATPase test kit. After the removal of medium, each well was added 200 µL of cell lysis buffer. After disassociation by repeatedly pipetting, cells were centrifugated at 12000 g for 5 min. The supernatant was preserved for further detection. Supernatant were tested with working fluid according to the kit procedure. Thus, luminance can be detected by fluorescence spectrophotometer (E6080, Promega Company). All ATP detection procedures were done at the temperature of 4 ℃.
Surgical procedure for spinal cord transection model and so-exo + pDA-Gel implantation
Rat SCI model was prepared through transection over spinal cord with a lesion about 4.0 ± 0.5 mm as previously mentioned24. Under deep anesthesia, hair on the back of the rats near their T10 spinous processes was shaved. With the separation of muscle, laminectomy was performed to expose spinal T9-T11 segments. A complete transection between the T9-T10 segment made a lesion gap of 4.0 ± 0.5 mm. So-exos + pDA-Gel was implanted between the gap of lesion site after hemostasis. SCI, pDA-Gel and rutin + pDA-Gel group were administrated in the same way to serve as groups for comparison. Animals receiving the same surgical procedure without spinal cord transections were set as sham group. Preparation of so-exo encapsulated gel is manipulated as previous experiment. In brief, so-exos were pipette into gels and incubated overnight. After the implantation, muscle and skin were separately sutured. Penicillin was intermuscular injected within 7 days after the surgical procedure. For better prognosis and restoration of urinary function, manual bladder massage was conducted twice daily until the micturition reflex recovery.
Locomotor function investigation
The postsurgical analysis of locomotor behavior was valuated weekly. Unrestrainedly, animals were moving in an open field, and were graded according to the 21-point Basso, Beattie, Bresnahan (BBB) locomotion rating scale by observers blinded to treatment group for each animal. Since all graded videos of rats were recorded, observers can repeatedly review the moving condition of each rat.
Sacrification for tissue, Hematoxylin and eosin (HE) and immunohistochemistry staining
Rats were sacrificed at the end of treatment experiment (Day 28) under deep anesthesia. Whole body were firstly perfused by isotonic physiological saline and then by 4% paraformaldehyde in PBS. Spinal cords surrounding the lesion site as well as major organs (hearts, livers, spleens, kidneys and bladders) were collected. The harvested tissues from each group were dehydrated in processing cassettes through a series of alcohol gradient overnight. After embedding, the section slices were stained according to HE staining kit. For immunofluorescence staining of the spinal cords, tissues were embedded and cryosectioned into 20 µm slices. After permeabilizing with Triton X100, sliced tissues were incubated with antibodies as neurofilament (NF) (Cell Signaling Technology, USA), glial fibrillary acidic protein (GFAP) (Boster, China), 4-hydroxynonenal (4-HNE) (Omnimabs, USA), 8-hydroxy-2’-deoxyguanosine (8-OHdG) (Omnimabs, USA) and choline acetyl transferase (ChAT) (Omnimabs, USA) overnight at 4 ℃. After rinsing with primary antibodies, the sections were incubated with Alexa Fluor 488- and 594-conjugated secondary antibodies (Jackson ImmunoResearch, USA) at 37 ℃ for 30 min. Nuclei were labeled via DAPI staining (Invitrogen, USA). The sections were observed using Olympus SLIDEVIEW VS200 (VS200, Olympus, USA).
Statistical Analysis
The quantitative results were shown as mean ± standard deviation values. Statistical analysis was calculated through GraphPad prism 8.4. Two-tailed unpaired t-test and one-way ANOVA were used under normal distribution and variance homogeneity. Significance can be proven by a P-value lower than 0.05.
{kind=link}