We report here the Saudi result from a multi-center study of a cohort of 21 patients with confirmed DADA2, including the update of previously reported 6 cases [18, 22, 27, 28]. Disease onset in our cohort remained in childhood, similar to the literature report. The mean age at presentation was 3.6 years, and 95% were less than 10 years. About 30% of our cohort presented before the first year of life. Indeed, the presentation of 4 patients (B1, B2, E2, J, and M) at birth or soon after birth emphasize the importance of considering DADA2 in patients with suspected clinical manifestation at such age. Only one patient (B4) presented with cutaneous vasculitis and cytopenia in adulthood. Presentation later in adulthood or asymptomatic individuals with DADA2 are well described in several reports. Indeed, patients with a predominance of the vascular phenotype are increasingly diagnosed by adult care providers [21, 29–31]. Molecular genetic testing and ADA2 enzyme levels are the diagnostic tools for DADA2. However, measurement of ADA2 enzyme activity may not be available in clinical laboratories, which made molecular genetic testing widely used [3, 10]. Diagnostic delays of DADA2 patients are observed in our cases. Time to diagnosis was prolonged with a mean of 3.9 years and may reach 11 years. Variability of phenotype, the rarity of the disease, limited availability of ADA2 enzyme testing, and poor awareness of DADA2 among physicians may impede consideration of this disease in the differential diagnosis with subsequent diagnostic delay. The establishment of a consensus diagnostic criteria for DADA2 may minimize such delay [32, 33]. Furthermore, the diagnosis of two (E2 and M) of our cohort after death highlight the need to enquire about unexplained deaths in the family on evaluating a patient with suspected DADA2.
The study sample is much less in number; however, in comparison to the analysis of 160 patients with DADA2 reported from the USA, Europe, and Turkey in one article [10], our cases are different in some clinical manifestations, including the frequency of phenotypes and genetic mutations (Fig. 2). The hematological and immunological manifestations predominate our cohort. Skin vasculitis and autoinflammatory features such as fever are less frequent at home than abroad. However, stroke is consistent in both cohorts. The frequency of cytopenias and impaired innate immunity are higher in our cohort. Tested ADA2 enzyme levels in our patients showed activity loss that coincides with their predominance of hematological phenotype [16].
Other rare clinical features, including growth retardation and complicated gastrointestinal ischemia, were reported in our cohort too. However, growth retardation, rigorously described in recent DADA reports, is found to impact a significant number (30%) of our patients [34]. Vasculitis manifestations of the gastrointestinal system generally include abdominal pain and, on rare occasions, intestinal necrosis with subsequent bowel perforation [12] Patient I, who presented with vasculitis phenotype, was noticed to have lower limb weakness that mimics transverse myelitis, which has not been previously described in children with DADA2.
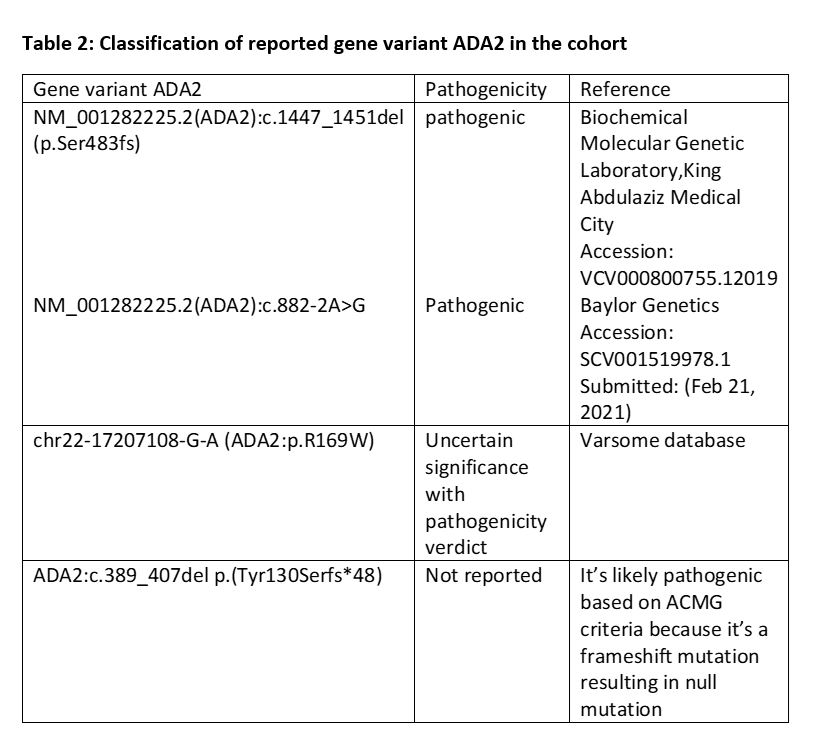
Distinct ADA2 mutations are reported in different ethnicities. For instance, the most frequent mutations in affected individuals are p.Gly47Arg in Turkish and Georgian-Jewish ancestry and p.Arg169Gln in Northern Europe population [5, 6, 7] ADA2 c1447-1451del and c882-2A: G are the most common identified mutations in 38% and 29% of our patients, respectively. G47R mutation was detected in one patient (I) with the predominance of vasculitis phenotype. One patient carried the R169Q mutation. Over half of DADA2 patients in the literature have homozygous ADA2 mutations [35]. While all our cohort but one are homozygous for ADA2 mutations and more than half of the patients had a family history of the disease (Table 1). The rate of consanguinity in the Saudi population was estimated in a large genetic study to be at 57%. [36] Thus, it is prudent to clarify the genetic status of relatives at risk and commence therapy accordingly to prevent complications.
Upon reviewing the ACMG variant classification 2015 and Varsome database, c.1447_1451del variant results in an amino acid change leading to an outframe deletion as delineated p. (Ser483Profs*5) and subsequently deleterious impact on the final protein function as well as structure which became evident by the complete enzymatic deficiency [37]. Thus, this variant is classified as pathogenic. The other variant, c.882-2A > G, disrupts the splicing of the final protein. Furthermore, the undetectable enzyme level has placed c.882-2A > G in the pathogenic classification. C has compound heterozygous variants, c.389_407del p.(Tyr130Serfs*48) and c.505C > T p.(Arg169Trp), the first variant yields a significant deletion of 19 nucleotides and outframe shift deletion resulting in a stop codon; this variant was not seen in any database including OMIM and to be classified as a likely pathogenic based on ACMG guidelines 2015. The latter one, c.505C > T p.(Arg169Trp), has been a classified variant of unknown significance, yet it has pathogenicity features. This sequence change replaces arginine with tryptophan at codon 169 of the ADA2 protein (p.Arg169Trp). The arginine residue is highly conserved, and there is a moderate physicochemical difference between arginine and tryptophan. This variant is not present in population databases. Additionally, c.505C > T disrupts the p.Arg169 amino acid residue in ADA2. Other variants (s) that disrupt this residue have been pathogenic (PMID: 24552285, 26867732, 24552284, and 25888558). These suggest that this residue is clinically significant and that variants that disrupt this residue are likely to be disease-causing Table 2.
The diverse clinical manifestation of DADA2 is observed in our patients, even among the same family. For instance, A1, despite multiple therapeutic trials, continues to suffer from severe disease due to transfusion-dependent cytopenias and recurrent infections, while his brother (A2) remained asymptomatic with no treatment. Springer et al. reported an adult patient with DADA2 who suffered from HL during early childhood [17]. Our previous studies reported three cases (A1, A2, and L) of HL associated with DADA2 with a novel deleterious mutation in the ADA2 gene [22, 28]. In the current study, we report an additional case of HL in (B3) the symptomatic carrier (single deleterious pathogenic ADA2 variant: c.882-2A: G). The observed association between HL and DADA2 in B3 adds additional cases in the literature and strengthens our team's reported data. B3 shows that in addition to the association between HL and DADA2, carriers of DADA2 could clinically and complication-wise behave as homozygous patients [11, 32]. This association between DADA2 and HL should alert the hemato-oncologist to the possibility of DADA2 as an underlying diagnosis in childhood HL, particularly in FHL and in the HL patient with an aberrant course and additional symptoms/signs as it is potentially essential both for genetic counseling as well as for optimal treatment.
The literature's most frequent clinical manifestations of DADA2 are related to small and medium arterial vasculopathy. Vasculitis most characteristically involves the central nervous system. Vasculopathy of the CNS manifests as early-onset (i.e., age less than 10 years) lacunar and/ or hemorrhagic strokes in one-third of DADA2 patients [3, 10]. Ischemic strokes are often small and deep in the brain stem, basal ganglia, thalamus, and internal capsule. Consequently, such strokes might not be evident in brain MRI or often manifest as silent strokes. Most individuals with abnormal brain MRI in our series had a silent ischemic stroke. Even without overt neurological manifestations, early detection of stroke by MRI brain and initiation of anti-TNF is recommended to prevent the accumulation of recurrent small infarcts that can lead to severe neurological impairment such as cranial nerve palsies, dysarthria, ataxia, and cognitive dysfunction [17].
Anti-TNF is the main current therapy in our cohort. TNF-inhibition in our cases is effective in preventing additional strokes [23]. However, for patients with a severe hematological phenotype including bone marrow failure and PRCA, TNF-inhibitor failed to reverse the hematological manifestations indicating the need for HSCT. Indeed, the most common reported indication for HSCT in DADA2 patients is cytopenia and or immunodeficiency, not responding to anti-TNF treatment [24, 25]. A1 suffered from corticosteroid-dependent PRCA and anti-TNF refractory cytopenia that responded to splenectomy, cyclosporine, and monthly IVIG. The role of splenectomy in treating cytopenia in patients with DADA2 is unclear; however, to date, there is insufficient data to establish risk-versus-benefit comparison definitively [38].
{kind=link}