Cell culture
Rat pheochromocytoma PC12 cells and Human Embryonic Kidney 293A (HEK293A) cells were purchased from the Chinese Academy of Sciences. The PC12 cells were cultured in 1640 medium (Gibco, New York, USA) supplemented with 10% horse serum (HS, Biological Industries, NA, USA) and 5% fetal bovine serum (FBS, Gibco, New York, USA). On the other hand, the HEK293A cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, New York, USA) supplemented with 10% FBS. All cells were cultured with 100 U/mL penicillin and 100 µg/mL streptomycin (P/S) (Solarbio, Beijing, China) at 37°C in a humidified incubator with 5% CO2 (Thermo Fisher Scientific, MA, USA).
Cell Viability
8×103 per well PC12 cells were seeded on 96-well plates (Thermo Fisher Scientific, MA, USA) and incubated with different concentrations of ultrasound-treated α-Syn PFFs (0, 5, 10, 20 and 50 µg/mL) for 24 h[35]. Thereafter, PC12 cell viability was measured using the Cell Counting Kit-8 (CCK8, Dojindo Laboratories, Kumamoto, Japan) following the manufacturer’s instructions. Briefly, at the end of treatment, 100 µL of media containing 10 µL CCK8 was added to each well and incubated in the 5% CO2 incubator at 37°C for 3 h. The optical density of each well was measured at 450 nm using Max F5 microplate reader (Molecular Devices, CA, USA). The experiment was repeated three times with three replicates in each treatment group.
Lrp1 Gene Knock Down
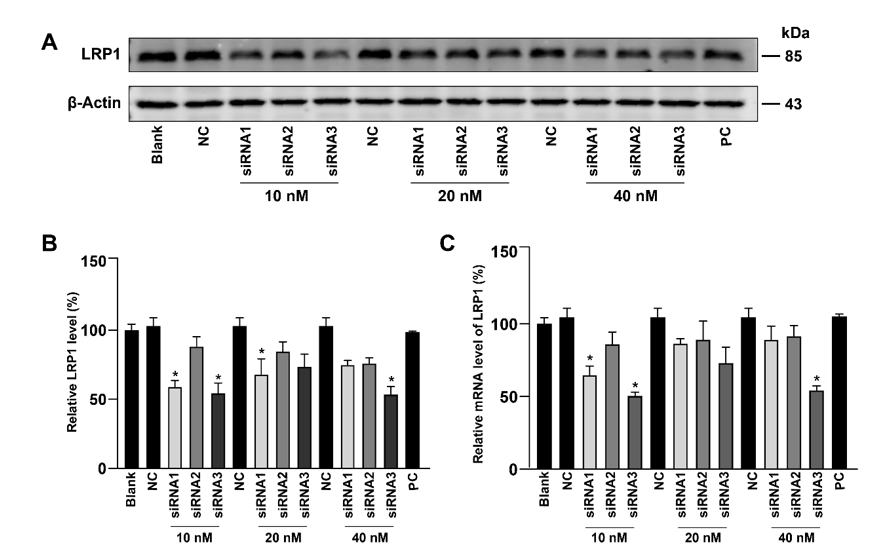
LRP1 siRNA (sequence of CCGAAATCTGTTCTGGACCAGTTAT), negative control (NC) and positive control (PC) siRNA were purchased from Invitrogen (Thermo Fisher Scientific, MA, USA). The PC12 cells at a density of 5 × 104 cells per square centimeter were cultured overnight in 6-well culture dishes (Thermo Fisher Scientific, MA, USA). The cells were then transfected with siRNA using Lipofectamine RNAi MAX transfection reagent (Thermo Fisher Scientific, MA, USA), following the manufacturer’s protocol. The efficiency of interference was detected using western blot and real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR).
Isolation Of Total Rna And Qrt-pcr
Total RNA was isolated from the PC12 cells using the RNAsimple Total RNA Kit (Tiangen, Beijing, China), and then cDNA was synthesized using TIANScript II RT Kit (Tiangen, Beijing, China), as instructed by the manufacturer. Afterwards, the samples were mixed with SYBR Green dye (Applied Biosystems, CA, USA), primers (Sangon Biotech, Shanghai, China) and RNase-free ddH2O for qRT-PCR. The primer sequences for LRP1, SNCA and GAPDH amplification were as listed in Table 1. The qRT-PCR experiment was performed on an ABI Fast 7500 system (Applied Biosystems, CA, USA). Relative quantification of mRNA expression was determined using the ∆∆Ct method. The LRP1 or α-Syn mRNA expression was normalized to that of GAPDH.
Table 1
Forward and reverse primer sequences for qRT-PCR
| Gene | Forward primer (5′-3′) | Reverse primer (5′-3′) |
| LRP1 | 5′-GGCTCTGTGGCTCAAAGGTA-3′ | 5′-CCTAAAAGTGGGAGCTGGGG-3′ |
| SNCA | 5′-CCTCCAACATTTGTCACTTGC-3′ | 5′-AGCAGGAAAGACAAAAGAGGG-3′ |
| GAPDH | 5′-GTTACCAGGGCTGCCTTCTCTTG-3′ | 5′-CCTTGACTGTGCCGTTGAACTTG-3′ |
Capping Lysine Residues On α-syn Pffs
Ultrasound-treated α-Syn PFFs were incubated with Sulfo-NHS-Acetate (NHS, Thermo Fisher Scientific, MA, USA) for 1 h to cap the lysine residues. 10 µg/mL of the lysine capped α-Syn PFFs were incubated with PC12 cells. After 24 h, the cells were harvested or fixed for western blot, qRT-PCR and immunofluorescence experiments.
α-syn Plasmid Transfection
Flag-tagged human full length α-Syn plasmid (SNCA), α-Syn N-terminus plasmid (N-SNCA) and α-Syn without N-terminus plasmid (ΔN-SNCA) in Escherichia coli were purchased from Sangon (Sangon Biotech, Shanghai, China). The HEK293A cells were pre-seeded on 6-well culture dishes with 1×105 cells per well to 70% confluency and then changed with fresh medium. Lipofectamine TM 3000 regent (Invitrogen, MA, USA) was used to coat and transfect 2 µg of the α-Syn plasmids into the cells. After 48 h, the HEK293A cells were harvested for western blot and immunofluorescence analyses.
Animals And Treatment
C57BL/6 male mice (4-5weeks) weighting 18–22 g were provided by Hunan SJA Laboratory Animal Co., Ltd, China. The mice were given free access to a standard laboratory chow and water, and were maintained in relative humidity and temperature, under a 12-hour light/dark cycle. The animal experiments were approved by the Ethics Committee of the Guilin Medical University (approval No. 2019-0021), and were performed according to the Institutional Animal Guidelines of Guilin Medical University.
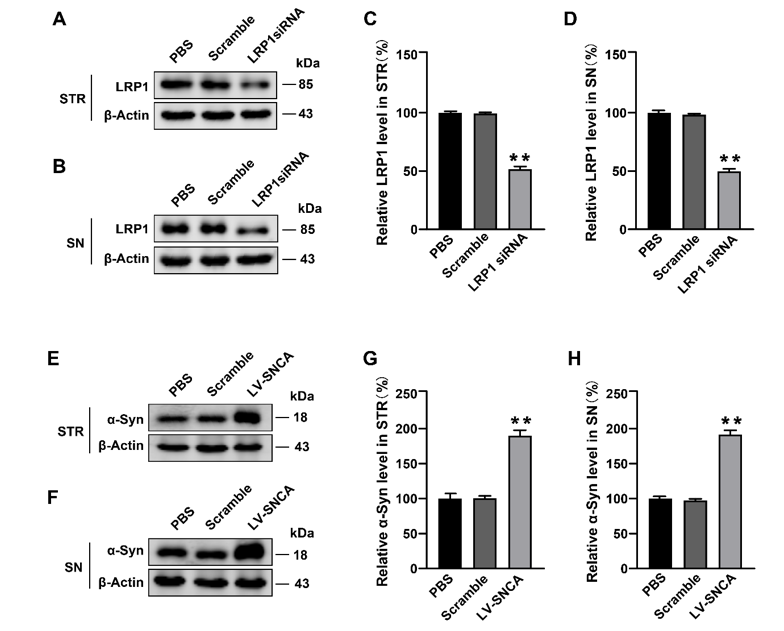
6 weeks old mice were randomly divided into PD model group (PFF group) and sham operation group (sham group), with 6 mice in each group. The PD model was established by injecting 2 µL of 1 µg/µL α-Syn PFFs into the right striatum (STR)[36, 16], at AP − 0.5 mm, ML − 2 mm, DV − 3 mm coordinates (relative to Bregma). The injections were performed under standard aseptic surgery conditions. A Hamilton needle was used to inject the α-Syn PFFs into the PD mice while equal volumes of 0.01 M phosphate buffered solution (PBS) were administered into the sham group mice at 0.5 µL/min. After operation, the mice were sterilized, sutured and kept quietly for 4 weeks.
On the other hand, lentivirus encoding either the siRNA targeting LRP1 or a control ‘scramble’ siRNA was injected into the 6-week-old wild-type mice through the tail vein. Eight weeks later, the human-α-Syn lentivirus was stereotactically administered into the STR. After six weeks, the mice were euthanized and then the brain tissues were excised for immunofluorescence and western blot analyses.
Mice Visual Gait Analysis
Visual Gait Analysis System (Xinruan Information Technology, Shanghai, China) was used to evaluate mice movement disorder[37]. The mice were trained for 3 days before the experiment. During detection, the mice were allowed to freely pass through the detection channel of a set length for at least three trials. The internal light source footprint refraction technology was used to perform efficient computer processing on the footprints in a video, and the movement of the mice were evaluated under natural walking. The tests were conducted at concentrated hours during the day while the channel was cleaned with 75% ethanol between the trials to wipe urine and feces to reduce interindividual interference.
Immunohistochemistry And Immunofluorescence
The mice were perfused with 0.9% saline followed by 4% paraformaldehyde[38]. Mice brains were stripped and further fixed for 24 h before dehydration with sucrose. The brains were frozen, sliced into 20 µm thick sections and permeabilized overnight using 0.3% TritonX-100 in PBS (PBST). The sections were then blocked in 2% goat serum for 30 min followed by incubation with mouse tyrosine hydroxylase (TH) antibody (1:2000, Sigma, MO, USA) at 4°C overnight. The samples were processed by a mouse SPN kit (ZSGB-BIO, Beijing, China) and diaminobenzidine (DAB, ZSGB-BIO, Beijing, China) solution. Thereafter, they were dehydrated and mounted and then imaged by a light microscope.
For immunofluorescence assays, the cells on coverslips were washed three times with 0.01 M PBS and fixed with 4% paraformaldehyde for 30 min at room temperature (RT). The cells were then permeabilized with 0.1% PBST for 20 min and blocked with 5% FBS. The cells and brain sections were incubated with the mouse antibody for α-Syn (1:1000, BD Transduction Laboratories, NJ, USA), rabbit antibody for LRP1 (1:2000, Beyotime Biotechnology, Shanghai, China) or mouse antibody for Flag (1:2000, Abmart, Shanghai, China) at 4°C overnight. Thereafter, the samples were incubated with a secondary antibody conjugated to Alexa Fluor 488 or 594 (1:2000, Invitrogen, MA, USA) for 1 h in RT. 4’, 6-diamidino-2-phenylindole (DAPI, 1:5000, Invitrogen, MA, USA) was used to stain the nucleus before mounting and then scanning with a confocal microscope (ZEISS, Oberkochen, Germany).
Western Blot
The cells and brain tissues were homogenized and lysed in RIPA lysis solution (Solarbio, Beijing, China), supplemented with protease and phosphatase inhibitor cocktails (Millipore, MA, USA) for 30 min. The samples were then centrifuged for 30 min at 12,000 g. The concentration of the supernatant was analyzed using the BCA method before mixing with 5 × loading buffer (Solarbio, Beijing, China). The samples were boiled for 10 min, and then 30 µg of total protein was loaded and separated by a 15% sodium dodecyl sulfate polyacrylamide gel. The samples were transferred to polyvinylidene difluoride membranes (Millipore, MA, USA) and blocked with 5% skimmed milk for 2 h. The membranes were then incubated with rabbit LRP1 antibody (1:5000, Beyotime Biotechnology, Shanghai, China), mouse α-Syn antibody (1:1000, Abcam, Cambridge, UK), mouse Flag antibody (1:2000, Abmart, Shanghai, China), mouse TH antibody (1:2000, Sigma, MO, USA), or mouse β-Actin antibody (1:10,000, Affinity, Melbourne, Australia) overnight at 4℃, followed by incubation with a fluorescent secondary antibody (1:10,000, LI-COR Biosciences, NE, USA) for 1 h. The blots were analyzed by ODYSSEY dual-color infrared fluorescence imaging system (LI-COR Biosciences, NE, USA).
Statistical Analysis
Data were presented as means ± SD, while GraphPad Prism 8.0 software (GraphPad, CA, USA) was used for statistical analyses. Comparison between the two groups was analyzed by unpaired t test. And comparison between three or more groups was analyzed by one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test. A P < 0.05 was considered statistically significant.
{kind=link}
{kind=link}