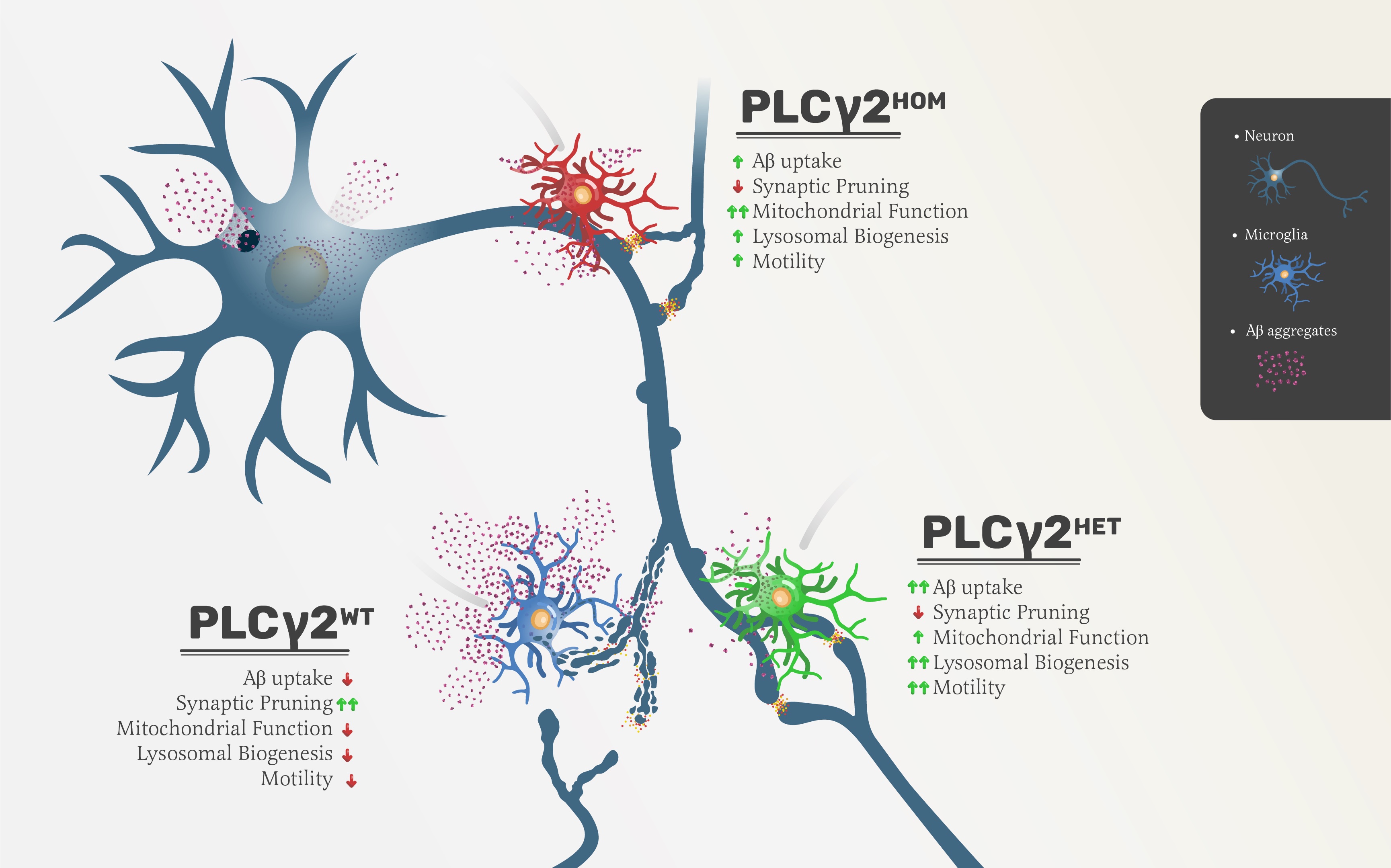
iPSC line and maintenance
hiPSC line BIONi10-C (Bio Sample ID: SAMEA3158050, ECACC ID:66540023) was purchased from EBiSC, which was deposited by Bioneer, and are available for research from European Collection of Authenticated Cell Cultures (ECACC). The PLCγ2 WT locus was authenticated through sequencing (Supplementary Table 1) and the PLCγ2HET and PLCγ2HOM hiPSC lines were generated on the parent BIONi10-C WT background inhouse using CRISPR editing. hiPSCs were cultured on Geltrex coated (100X) (A1413202, Gibco) plates and maintained at 37oC and 5% CO2 in E8-Flex medium (A2858501, ThermoFisher).
CRISPR editing
In brief, to insert the P522R variant, BIONi010-C WT hiPSCs were dissociated to single cells using StemProTM AccutaseTM (A1110501, ThermoFisher). Cells were then seeded at 5X105 per well of a 6-well plate and incubated for 3-4 hrs to allow for attachment. sgRNA complex was prepared by mixing 100 µM stock of trcrRNA (1072532, IDT) and crRNA (sequence – CCTACAGAACTACATTTTGG), targeting exon 17 of the PLCγ2 locus, in equimolar concentration with 196 µL of nuclease free duplex-buffer (IDT) to a final volume of 200 mL and annealing at 95oC for 5 minutes. At the same time, 1 µM stock Cas9 RNP was made by adding 1 µL of 62 µM Hi-Fi Cas9 RNP (1081060, IDT) to 61 µL of Opti-MEMTM (31985070, ThermoFisher). 12 µL of sgRNA complex and 12 µL of Cas9 RNP were then mixed in 76 µL of Opti-MEM and incubate at room temperature for 5 min. In parallel, 4 µL of LipofectamineTM Stem (STEM00008, ThermoFisher) and 96 µL of Opti-MEM were mixed and incubated for 10 min at room temperature to make the transfection mixture. 2 µg of donor plasmid and 500 ng of puromycin selection construct were added to the sgRNA:Cas9 (RNP) complex and transfection mixture to a final volume of 200 µL, and incubated for 10 mins. The medium on the plated cells was replaced with 2 mL of pre-warmed Opti-MEM supplemented with RevitaCellTM (100X) (A2644501, ThermoFisher) and transfection mix was added on top in a drop-wise manner before mixing gently through swirling the plate. Cells were left for four hours in the incubator before medium top-up with E8-flex supplemented with RevitaCell and then being left on overnight. Medium changes were performed until cells reached 70-80% confluency, whereafter, single cell selection with 0.25 mM Puromycin (A1113803, ThermoFisher) was done to isolate pure colonies. Single cell colonies were established, expanded and banked. To confirm conversion of 522P to 522R, DNA was extracted using DNeasy Blood & Tissue (69506, QIAGEN) and sent for sequencing (Eurofins Genomics) (sequencing primers are available in supplementary methods Table 1) and positive colonies were identified to be used in downstream assays.
Generation of iPSC derived microglia
Microglia-like cells were generated following Haenseler et al. (Haenseler et al., 2017). In brief, embryonic bodies (EBs) were formed through plating and spinning of 3X106 BIONi010-C (BINI-10) (WT, PLCγ2HET and PLCγ2HOM) at 300 g on an AggreWell 800 plate (34850, StemCell Technologies) in E8-Flex medium supplemented with 50 ng/ml BMP4 (120-05ET, Peprotech), 50 ng/ml VEGF (PHC9394, ThermoFisher) and 20 ng/ml SCF (300-07, PeproTech). 75% medium change per day was performed for 72 hours, after which EBs were transferred to a T75 flask and maintained in X-VIVO15 (BE02-060F, Lonza) supplemented with 25 ng/ml IL-3 (PHC0031, ThermoFisher), 100 ng/ml M-CSF (300-25, PeproTech), 2 mM Glutamax (35050061, ThermoFisher) and 0.055 mM β-mercaptoethanol (31350-010, ThermoFisher). Medium was topped up every week and after 4 weeks, emerging precursor cells were collected and differentiated to microglia-like cells for 7-days in microglia medium consisting of 25 mL DMEM/F12 (11330032, ThermoFisher) and 25 mL Neurobasal Plus media (A3582901, ThermoFisher) supplemented with 100 ng/ml M-CSF (300-25, Peprotech), 100 ng/ml IL-34 (200-34, peprotech) and 10 ng/ml GM-CSF (300-03, Peprotech), 2 mM Glutamax (35050061, ThermoFisher), 0.05 5mM β-mercaptoethanol (31350, Life Technologies), and 0.25 mg/mL Insulin (I9278, Sigma).
Generation of iPSC derived cortical neurons
iPSC derived cortical neurons differentiation was adapted from Fernandopulle et al. (Fernandopulle et al., 2018), with slight modifications. In brief, BIONi010-C (BINI-10) iPSC cell lines (PLCγ2HET) were dissociated using accutase and seeded as single cells at a density of 5X105 per well of a six-well plate. The cells were left to attach for 4 hrs. Cells were then transduced in a similar manner to the above protocol of PLCγ2 editing, but in this case using a CRISPR-cas9 RNP to stably integrate a doxycycline inducible NGN2 expression cassette (see supplementary methods - plasmids), into the CLYBL safe harbour site, allowing for the driving of cellular differentiation into cortical neurons. Double selection was performed using Blasticidine (100ng/mL) (Code) and mApple markers to identify positive colonies. iPSCs stably expressing NGN2 were then induced in DMEM/F12 with HEPES (11330032, ThermoFisher) supplemented with; N2 (1X) (17502048, ThermoFisher), NEAA (1X) (11140050, ThermoFisher), Glutamax (1X) (35050038, ThermoFisher) and doxycycline (2mg/ml) (D9891, Sigma). 72 hours post induction, the cells were transferred to P-D-L (product code) and Laminin (1:100 dilution in PBS) (23017015, ThermoFisher) coated plates and fed with Neurobasal Plus media (A3582901, ThermoFisher) supplemented with; B27 (50X) (17504044, ThermoFisher), 10 ug/ml BDNF (450-02, PeproTech), 10 ug/ml NT-3 (450-03, PeproTech) and 1 mg/mL Laminin for up to 12 days.
BV2 Culture
BV2 cells were cultured and maintained at 37°C, 95% O2 and 5% CO2 in Dulbecco’s modified Eagle’s medium (31885-023, Gibco) supplemented with 10% (v/v) foetal calf serum (A4768801, ThermoFisher) and 1% pen/strep (15140122, ThermoFisher).
Overexpression of PLCγ2 in BV2 cell line
The overexpression construct PLCγ2P522R point mutation was inserted into the human PLCγ2WT plasmid (RC200442, Origene) using Q5® site directed mutagenesis kit (E0554S, NEB) according to manufacturer’s protocol. The mutagenesis primers used are listed in supplementary Table 1. For overexpression experiments, in brief BV2 cells were seeded onto either a 12 well plate or 8-well chamber slide (IB-80827, iBIDI) at a density of 140,000 and 21,000 cells per well respectively and allowed to settle for 24 hrs. 2 hrs prior to transfection a complete media change was done with fresh DMEM without pen/strep. For transfection, 50 hg/100 ml of cDNAs (Control, PLCγ2WT and PLCγ2P522R plasmid) were mixed with 25 ml/100 ml Opti-MEMâ and 0.27 ml/100 ml of LipofectamineTM 2000 (ThermoFisher) and incubated for 30 minutes. Afterwards, the media on the cells was replaced with transfection mix and was incubated for 24 hrs. The media was then exchanged for fresh DMEM, and cells were incubated for a further 24 hrs before downstream assays, including uptake and metabolic profiling.
Protein extraction and immunoblotting
Protein was extracted from cultured BV2 cells or 7 day mature iPSC derived microglia by lysis for 30 min at 40C in ice cold RIPA buffer (150 mM NaCl, 10 mM Tris, pH 7.2, 0.1% SDS, 1% Triton X-100, 1% deoxycholate, 5 mM EDTA), supplemented with protease (78430, ThermoFisher) and phosphatase inhibitor (524625-1SET, MERCK) cocktails (ThermoFisher). The RIPA soluble fraction was obtained by centrifugation of the samples at 10’000 xg for 10 minutes. Protein concentration was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Samples were mixed with loading buffer (928-40004, Li-Cor) before boiling at 950C. SDS-PAGE separation was performed on NuPAGE 4–12% gradient Bis-Tris gels (Thermo Fisher) and proteins were transferred to nitrocellulose membranes (IB23002, ThermoFisher) using an iBLOT2 following manufactures protocol. Membranes were blocked for 1 hour in 3% BSA in TBST (0.1% tween) before incubation with primary antibodies (supplementary Table 2) in 3% BSA in TBST overnight at 40C. Membranes were washed 3 times 5 minutes and incubated with secondary antibodies (supplementary Table 2) for 1hr at room temperature before further washes using TBST. Membranes were analysed using a LI-COR Odyssey CLx system and protein levels were quantified using Image Studio Lite (LI-COR Biosciences), normalized to their corresponding b-Actin levels. Statistical significance was assessed using one-way ANOVA.
Phagocytosis assay
For phagocytosis assay 5 x 104 iMG progenitors were seeded onto 8-well chamber slide (IB-80827, iBIDI) coated with PDL and matured for 7 days with 50% medium change every other day. Uptake assays were then performed by exchanging 100% media to fresh media supplemented with either 250 nM Ab1-42 HyLite Fluor 647 (AS-60493, AnaSpec), mouse brain purified tdTomato-tagged synaptosomes generated in-house, FITC-Dextran 4kD (46944, Sigma), FITC-Dextran 150 kD (46946, Sigma), Zymosan beads-568 (Z23374, ThermoFisher) and 75 nM LysoTracker DND-99 (L7528, ThermoFisher) for 100 min. After elapsed time, iMG were washed with PBS and fixed with 4% paraformadehyde for 30 min. Fixed iMG were subsequently stained with SNL (FL-1301-2, Vector) to visualise the membrane and with 2 uM Hoechst (62249, ThermoFisher) nuclear stain. Images were acquired using Nikon A1R inverted confocal microscope (Nikon) running the NIS elements software using 60x oil immersion lens. Acquired images were analysed using FIJI and IMARIS, thresholding to WT/Control, marking ROI and quantified either as %Cell Area or uptake (cargo) volume per total cell volume. Statistical significance was assessed using a Kruskal-Wallis test.
Immunochemiluminiscence Assay (Meso Scale Discovery)
To measure the residual level of Ab in the co-culture assay, supernatant was collected from co-culture of 21-day mature iPSC derived cortical neurons and 7day mature microglia and centrifuged at 1000G to remove any cellular debris. In brief Immunochemiluminiscence assay was performed by adding 150 μL of Diluent 35 (MSD) to each well of a 96 well multi-spot MSD plate (N45197A-1, MSD) and incubated at room temperature for 1 hour, on a plate shaker. Subsequently, the plate was washed 3 times with Tris Wash Buffer (MSD). After washing, 25 μL of Ab detection antibody (6E10, MSD) and 25 μL of prepared samples (cellular supernatant) was added to each well. The plate was incubated at room temperature for 3h, on a plate shaker, prior to washing 4 times with Tris Wash Buffer. 150 μL of 2X Read Buffer (MSD) was added to each well. Samples were analysed on the MSD plate reader using default detection criteria. The MSD read outputs, indicative of Ab level, were normalized to their corresponding total protein concentration and were analysed using GraphPad. Statistical significance was assessed using a Kruskal-Wallis test.
RT-qPCR analysis
RNA was extracted from iPSC derived microglia (iMG) and/or BV2 using Direct-zol RNA MiniPrep Kit (Zymo research) according to the manufacturer’s protocol. For RT-qPCR analysis, RNA was reverse transcribed into cDNA using iScript cDNA synthesis kit (BIO-RAD) according to the manufacturer’s protocol. Target specific PCR primers for mouse and human (Supplementary Table 3) were obtained from IDT. For qRT-PCR analysis Takyon Rox SYBR MasterMix dTTP blue (Eurogentec) was used. For relative expression analysis the DDCt comparative method was used to compare TBP normalized expression level of the target mRNA. Data were normalized either to control (BV2) or WT groups (iMG) and are shown as median ± SD. Statistical significance for each gene was assessed using a Kruskal Wallis test.
Cellular respiration assay
To measure oxygen consumption rates (OCR) and extracellular acidification rates (ECAR) in real time, iMG were seeded and matured for 7-days on a Seahorse 96 well cell-culture microplate (PerkinElmer) at a density of 25’000 cells per well. Mature iMG’s mitochondrial respiration was then analysed on the seahorse XFe96 Analyser (Agilent Technologies) using a Mitostress test kit (Agilent Technologies) according to manufactures protocol.
For BV2 cells, to measure OCR and ECAR, cells were seeded on a seahorse cell culture plate at a density of 27’000 cells per well and transfected, as described, with cDNA (Control, WT and PLCγ2-P522R) constructs. 24 hrs post transfection, real-time mitochondrial respiration was measured using a Mitochondrial stress test kit (Agilent Technologies) on the seahorse XFe96 Analyser. All data were analysed using Wave v2.4.0.6 (Agilent technologies) and Graph pad. Statistica significance was assessed using a Kruskal-Wallis test.
Cell tracking/motility assay
To measure microglia motility, speed, and distance, iMG were plated on a glass bottom Ultra cell carrier 96 well plate (6055302, PerkinElmer), precoated with PDL, and matured for 7 days as previously described. Mature iMG were stained with nuclear mask blue (ThermoFisher) and cell mask orange (ThermoFisher) for 30 min. Cells were then washed with PBS twice and 100 ml of prewarmed microglia media made in FluroBright (A1896701, Gibco) was added. iMGs were imaged for 2 hrs on a high-throughput imaging OperaPhenix (PerkinElmer) with both temp and CO2 maintained at 37oC and 5% respectively. Image analysis was done using Harmony cell tracking software. Both tracked nuclear speed and cytoplasmic speed were exported and analysed using Graphpad.
Calcium imaging
To monitor Ca2+ levels in iMG, microglia progenitors were seeded on a glass bottom cell carrier Ultra 96 well plate and glass bottom 8-well chamber ibidi slides, precoated with PDL. iMG were matured for 7 days following the described protocol. To treat cells, mature iMG were washed with PBS once and 100 ml FloBright microglia media supplemented with cell permeable 2 mM Fura Red™, AM, cell permeant (F3021, ThermoFisher) was added to the 96-well plate and 300 ml was added to the iBidi plates and incubated for 30 min. Cells were then washed with PBS and FloBright microglia media devoid of Fura Red was added. Images were taken on a high-throughput microscope OperaPhenix (PerkinElmer) and Nikon spinning disc (Nikon), with analysis being performed using Harmony software and ImageJ. Background was subtracted from each frame and ratio-metric value of bound to unbound Ca2+ was extracted and analysed in GrapPad.
{kind=link}