Characterization of LPPC-DOX, LPPC-CpG and LPPC-DOX-CpG
First, the capacities of LPPC for DOX encapsulation were determined. LPPC (1 mg) exhibited a maximum DOX encapsulation of approximately 0.5 mg (Table 1). In addition, the encapsulation of DOX by the LPPC did not significantly alter the average zeta potential and only slightly increased the particle size by a factor dependent on the DOX increase (Table 1). In conclusion, the LPPC particle size and average zeta potential were approximately 210 nm and 42 mV, respectively.
Sequentially, drug release from LPPC-DOX complexes was assessed. During 120 h of incubation, kinetic analysis of drug release showed that only 20% of the encapsulated DOX was released from LPPC-DOX at 4 °C, while approximately 40% and 98% of the encapsulated DOX was released into the media at 25 °C and 37 °C, respectively (Fig. 1A). The results indicated that the release rate of DOX from the LPPC was temperature dependent.
Second, the CpG ODN absorbing capacity of LPPC or LPPC-DOX complexes was determined. As shown in Fig. 1B, 50 μg of the LPPC adsorbed approximately 9 μg of CpG, which was equal to the absorption ability of LPPC-DOX. Furthermore, CpG absorption led to an increase in the particle size (244.5±9.3 nm) but to a decrease in the zeta potential (29.2±2.9 mV) of LPPC-DOX complexes. Moreover, the drug release kinetics did not significant differ between LPPC-DOX and LPPC-DOX-CpG (Fig. 1C). Together, these results revealed that DOX encapsulation did not interfere with CpG binding and that the binding of CpG to LPPC-DOX did not influence the characteristics of drug release from LPPC particles.
Biofunction of LPPC-DOX-CpG in vitro
Compared to DOX, LPPC-DOX and LPPC-DOX-CpG had higher cytotoxic activities (IC50; 5.5- and 4.8-fold, respectively), and the LPPC obviously increased the cytotoxic effect of DOX on B16F10 cells (Fig. 2A). The cytotoxic kinetics revealed that CpG absorption on LPPC did not influence the cytotoxic activities of LPPC-DOX (Fig. 2A; green and purple lines).
Likewise, the adjuvant effects of LPPC-DOX-CpG complexes on cytokine induction were also examined. As shown in Fig. 2B, LPPC-CpG dramatically increased the expression of the IFN-a, TNF-a, and IL-6 cytokines in splenocytes compared to that induced by CpG alone, which indicated that the LPPC facilitated CpG to activate stronger immune responses. However, the LPPC induced only slight expression of these cytokines, which indicated that the LPPC and CpG had a synergistic effect on cytokine induction. Moreover, quantitative analysis revealed that compared to CpG alone, LPPC-CpG efficiently enhanced the mRNA expression of IFN-a by nearly 4-fold, TNF-a by 13-fold, and IL-6 by 11-fold (Fig. 2C).
LPPC-DOX-CpG administered transdermally exerts antitumor activity in vivo
Subsequently, the transdermal kinetics of LPPC encapsulated DOX were monitored, revealing that transdermal DOX was detectable within 12 h and undetectable after 24 to 36 h (Fig. 3A). According to the kinetic analysis results, the animals were treated once a day for the following animal study.
C57BL/6J mice bearing B16F10 tumors were transdermally treated with empty LPPC, LPPC-DOX, LPPC-CpG, or LPPC-DOX-CpG once every day. B16F10 tumor growth was significantly suppressed in animals treated with LPPC-DOX, LPPC-CpG, and LPPC-DOX-CpG compared with PBS (Fig. 3B). Compared to LPPC-DOX and LPPC-CpG, LPPC-DOX-CpG exerted a better tumor suppressive effect, it could cause significance in decrease of tumor growth from 32nd to 34th days after treatments. Both LPPC-DOX and LPPC-CpG effectively inhibited tumor growth, and the differences were not statistically significant. Interestingly, although the effect is not weaker, empty LPPC treatment also led to a significant anti-tumor effect on tumor growth comparing to PBS group from 18th to 29th days. These results showed that LPPC-DOX-CpG inhibited the growth of malignant B16F10 cells more effectively than LPPC-DOX and LPPC-CpG.
Moreover, the mice treated with LPPC-DOX-CpG also exhibited the longest survival time (T1/2=47.5 days); LPPC-DOX and LPPC-CpG extended the survival times (T1/2=40 days and 40 days) compared to those in the control groups (PBS and LPPC, and T1/2=26.3 days and 35 days). Interestingly, 20% of mice treated with LPPC-DOX-CpG were tumor free for more than 60 days, and 10% of mice treated with LPPC-DOX and LPPC-CpG were tumor free (Fig. 3C).
Furthermore, the pathological changes in tumor areas were histologically monitored after treatment with the different agents, revealing that LPPC-DOX-CpG significantly shattered the tumor nucleus and dramatically damaged the tumor tissue. Obviously, LPPC-DOX and LPPC-DOX-CpG also attacked the tumor vessel, resulting in bleeding in the tumor area (Fig. 3D). In addition, while all LPPC-related treatments induced the infiltration of immune cells, LPPC-DOX-CpG recruited the most immune cells to the tumor area (Fig. 3D). These results potentially indicate that the transdermal delivery of this LPPC system inhibited tumor growth by activating the immune response.
Pathologic effects of LPPC-DOX-CpG in vivo
The histopathological tumor-suppressive effects of transdermal LPPC-DOX-CpG treatment were further explored. LPPC-DOX-CpG reduced the expression of PCNA in tumor cells, indicating that tumor growth may have been partially suppressed by the inhibition of cell proliferation (Fig. 4). Moreover, LPPC-DOX and LPPC-CpG reduced the expression of PCNA in tumor cells, but their effects were weaker than LPPC-DOX-CpG. LPPC-DOX-CpG, LPPC-DOX and LPPC-CpG significantly increased the active caspase 3 compared with that in the LPPC and PBS groups. LPPC-DOX-CpG had the strongest ability to activate caspase 3 and induced the cell apoptosis better than LPPC-DOX and LPPC-CpG.
Moreover, tumor-infiltrating cells were also examined by IHC, revealing that the CD3, CD4 and CD8 antigen levels were increased in the tumor area after LPPC-DOX-CpG, LPPC-DOX and LPPC-CpG treatment. Thus, these treatments induced the infiltration of CD4T or CD8T lymphocytes into tumor areas, and the numbers of CD8T cells were slightly higher than those of CD4T cells (Fig. 4; CD3, CD4, CD8). Similarly, the expression of CD19, a B cell marker, was increased after LPPC-DOX-CpG, LPPC-DOX or LPPC-CpG treatment. Likewise, increased levels of the CD68, CD86 and CD163 antigens were observed in tumor areas, which indicated increased macrophage infiltration into the tumor area and differentiation into both the M1 and M2 subtypes (Fig. 4).
Systemic effect of transdermally administeredLPPC-DOX-CpG
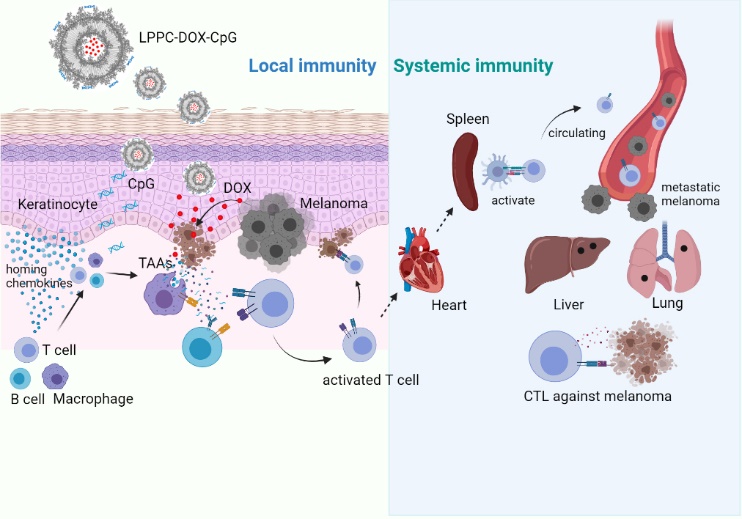
We next assessed whether the transdermal administered LPPC-DOX-CpG could induce systemic antitumor immunity. In previous in vivo therapy study, tumor-free mice were sacrificed, and the specific cytotoxicity of their splenocytes was examined. As shown in additional file 1, the splenocytes obtained from mice treated for 60 days exhibited antitumor activity, indicating that the transdermal administration of LPPC-DOX-CpG induced systemic immunity against melanoma cells.
In this model, the subcutaneous melanoma cells spontaneously metastasized to colonize the liver or lung and significantly formed colonies (Fig. 5A, PBS and LPPC treatments). These colonies preferred to colonize beside vessel, which suggested that they spread primarily through blood circulation. Transdermal administration of LPPC-DOX-CpG induced systemic immunity against melanoma cells, thereby reducing tumor cell metastasis. The effect of LPPC-DOX-CpG on the distant metastasis of melanoma cells was monitored, revealing that LPPC-DOX-CpG and LPPC-DOX significantly reduced the sizes and numbers of metastatic colonies in the liver and lung (Fig. 5B and 5C). Therefore, these results showed that LPPC-DOX-CpG administered transdermally not only suppressed subcutaneous melanoma growth but also reduced the metastasis of melanoma cells. Systemic antitumor immunity may have contributed to this result.
Molecular mechanisms underlying the therapeutic tumor-inhibitory effects of transdermal LPPC-DOX-CpG
Furthermore, the gene expressions in tumor samples of different groups respectively were analyzed by NGS. First, the genes with significantly different expressions in the samples treated with LPPC-DOX-CpG comparing to the treatment of PBS (p < 0.05) were selected. The selected genes with the fold changes (FCs) between 1.1 and 0.9, or the values of expressions (FPKM) less than 0.1 both were further excluded. In total, 468 genes met these criteria, and their names, ID, and parameters (including mean of expression, FC and p value) were displayed in additional file 2, respectively. Then, their functions were annotated with the GENECARD online database and divided into 27 categories (Fig. 6A). The gene expression values and FCs were shown in Fig. 6B. The genes were shown to be associated with 4 major functions: immune (22.6%), cell growth (15.4%), metastasis (7.3%) and apoptosis (4.9%), and their values of gene expressions and FC were labeled with color (Fig. 6B). The average expression levels and FCs as well as the distributions were shown in Fig. 6C and 6D. The genes in the metastasis group had the highest expression levels, while those in the immune group had the highest FC values. The major pathways in the four groups were analyzed as follows.
Molecular mechanisms underlying the therapeutic immune effects of transdermal LPPC-DOX-CpG
Additional file 3 showed 66 immune-non-enriched differential expression genes (DEGs) in the categorization of immunity were 52 up-regulations and 14 down-regulations for gene expression. And there are another 40 DEGs enriched in certain important pathways according to KEGG, GSEA, and published literature (Additional file 3).
Chemokine pathways: As shown in Fig. 7A, Pf4 and CCL2 (FC = 2.5 and 2.2) but not CXCL13 (FC = 0.8) were up-regulated and engaged their receptors including the increasing CCR5, and trigger the cell migration pathways including CCR5 pathway and GPCR pathway. In addition, a macrophage homing marker (CD68) and migration gene (ADGRE5) were increasing (FC = 1.84 and 1.7; Table S2) that indicated the chemo-attracting pathways were activated and cause the macrophages homing to the tumor area. It revealed that LPPC-DOX-CpG treatment could induce chemokine releases and attract the macrophage migration to tumor area through CCR5 and GPCR pathways. As shown in Fig. 7A, LPPC-DOX-CpG caused 10 DEG expressions, but LPPC-DOX or LPPC-CpG only induced 2 significant different expressions (not including CD68) in this pathway (p<0.05). Obviously, only LPPC-DOX-CpG could lead to such effect by inducing the CCL2 and Pf4 releases to attract the migration of immune cell through CCR5 or CXCR3/5 mediated pathway (Fig 7B), but not other treatments.
Interferon-g pathway: Although the expression of IFN-g did not significantly differ between the PBS and LPPC-DOX-CpG groups, the expression levels of 11 IFN-g-responsive genes (direct and indirect) were increased (Fig. 7C). Besides, MHC-relative gene expressions obviously were increased, in which 4 DEGs (CTSB, CTSL, CTSS and XBP1: Fc= 1.9, 1.6, 3.6 and 1.3) increased for MHC II expressions and three DEGs (LILRA6, TAP1 and TAPBP: Fc=1.1, 1.7 and 1.6) for MHC I expressions. Among the 11 DEGs related to INF-g (all which were upregulated by LPPC-DOX-CpG treatment), only one (Gbp7) was significantly upregulated by LPPC-CpG, and LPPC-DOX failed to upregulate any genes, indicating that both DOX and CpG were essential for activation of the INF-g pathway (Fig. 7D). These results indicated that LPPC-DOX-CpG, but not LPPC-DOX or LPPC-CpG, activated the INF-g pathway.
NF-kB pathway: Moreover, the expression levels of genes associated with the NF-kB pathway were increased after LPPC-DOX-CpG treatment. Ten upregulated DEGs and 1 downregulated DEG were enriched in the inflammatory pathway (Fig. 8A). TLR, IL1R, FcR and LT-BR triggered different signaling pathways to thereby activate NF-kB. Consequently, the levels of TLR-inducible gene ZC3H12A and NF-kB down-steam gene SRGN were also increased by 1.5- and 3.5-fold, respectively, which indicated that LPPC-DOX-CpG treatment activated the NF-kB pathway. Moreover, the expression of CEBPB, a marker of macrophage activation, was increased by 2.2-fold in the LPPC-DOX-CpG treatment group. In addition, LPPC-DOX and LPPC-CpG failed to activate this pathway, which revealed that both DOX and CpG were essential for its activation (Fig. 8B).
T- and B- cells: LPPC-DOX-CpG treatment also enhanced the migration and activation of T- and B-cells by significantly upregulating the expression of 13 DEGs (Fig. 8C). In addition, B-cell migration and activation were potentially activated by the increased expression of the IL-21 receptor pathway. The expression of CD24A, a marker of B-cell activation, was obviously increased in the LPPC-DOX-CpG group (FC = 1.9), indicating that LPPC-DOX-CpG induced B-cell activation. Among these DEGs, LPPC-CpG induced only one DEG and LPPC-DOX induced none, indicating that the LPPC-DOX-CpG-induced activation and migration of T- and B- cells required DOX and CpG (Fig. 8C).
Taken together, these data demonstrated that LPPC-DOX-CpG treatment would induce chemokine releases (Pf4 and CCL2) that would cause macrophage migration to tumor area through CCR5 and GPCR pathways, and activating IFN-g and NF-kB pathways. These signaling would consequently cause the increases in the expressions of MHC I and II, and result in enhancing the presentation and activation of macrophage. Sequentially, the activated macrophages would recruit the lymphocytes to infiltrate into tumor area and activate them.
Molecular mechanisms underlying the therapeutic effects of transdermal LPPC-DOX-CpG on tumor growth and metastasis
Cell proliferation: Based on the annotation of genes related to cell proliferation in the GENECARD online database, in total 72, there are 51 proliferation non-enriched DEGs (Additional file 4; 29 decreases and 22 increases in the expressions). Other 21 DEGs were analyzed by KEGG, revealing that were enriched in the RAS-related and Wnt pathways (Additional file 4, 9 upregulated and 12 downregulated). LPPC-DOX-CpG treatment mainly inhibited the H-ras/RAF and wnt/b-Catenin/TCF7L1 pathways but also partially enhanced the PI3K/AKT pathway, including the upstream genes EPO and APLN and the downstream gene MTOR (Fig. 9A). In addition, CDK2 expression was increased. Taken together, these results suggested that the inhibition of tumor proliferation by LPPC-DOX-CpG treatment was not the main mechanism influencing tumor growth, which was consistent with the IHC results (Fig. 4). Among the 21 DEGs enriched by LPPC-DOX-CpG treatment, only 9 were significant enriched by LPPC-CpG, and 5 were significantly enriched by LPPC-DOX (p<0.05, Fig. 9B). According to these results, LPPC-DOX-CpG requires both DOX and CpG to exert a minor inhibitory effect on cell proliferation.
Apoptosis: Fourteen genes related to apoptosis were upregulated, and 9 were downregulated (Additional file 5). These 23 DEGs were further analyzed, revealing that 16 were significantly enriched in several pathways associated with apoptosis. LPPC-DOX-CpG treatment activated the exogenous apoptosis pathway through the TNF receptor and Fas pathway (Fig. 10A). These pathways were promoted by the increased expression of caspase activation genes and the decreased expression of caspase inhibition genes. Furthermore, pro-survival genes such as Bcl-2 were downregulated. These results indicated that LPPC-DOX-CpG may have inhibited tumor growth by disrupting the balance between the expression of pro-apoptotic and anti-apoptotic genes to induce apoptosis through the extrinsic apoptotic pathway (Fig. 10A). Furthermore, LPPC-CpG induced the significant differential expression of only 1 gene, and LPPC-DOX induced the significant differential expression of 4 genes (Fig. 10B). These results revealed that LPPC-DOX-CpG required the synergistic effect of DOX and CpG to efficiently trigger apoptotic reactions.
Metastasis: Thirty-two DEGs involved in tumor metastasis by LPPC-DOX-CpG treatment, of which 16 were downregulated and 16 were upregulated (Additional file 6). After analyzing their functions, 14 DEGs were found to be enriched in metastasis pathways, suggesting that LPPC-DOX-CpG treatment reduced tumor metastasis by downregulating metastasis-related genes and increasing the expressions of metastasis-inhibitory genes. LPPC-DOX-CpG treatment mainly reduced the expressions of genes associated with cell adhesion to thereby downregulate RhoA-related pathways and increased the gene expressions, including ARHGAP23, ARHGAP25 and RND3, to inhibit RhoA pathways (Fig. 11A). In addition, LPPC-DOX-CpG treatment also down-regulated the Smad2/3-mediated pathway to decrease EMT. LPPC-CpG induced the significant differential expression of 3 genes, while LPPC-DOX-CpG induced the significant differential expression of 14 genes (Fig. 11B) (p<0.05), and LPPC-DOX induced the significant differential expression of only 1 gene. These results indicated that the anti-metastatic ability of LPPC-DOX-CpG required the synergistic effect of CpG and DOX.
Together, these NGS results revealed that the transdermal LPPC-DOX-CpG successfully activated host local immunity to induce tumor cell apoptosis and exerted a systemic antitumor effect to decrease tumor metastasis. Thus, local LPCC treatment simultaneously induces tumor cell death and activates immunity and may be a safe and efficient therapeutic strategy for skin-associated cancers.
{kind=link}