To investigate the diversity and prevalence of viruses in natural ecosystems, we sampled 32 wild citrus trees in the Ailao Mountains, an area that harbors a very diverse flora [15] and is located within the region where citrus plants originated [10]. Some viruses may have coevolved with these wild hosts over a long period of time, making the latter natural reservoirs for viruses that could cause new or re-emerging diseases.
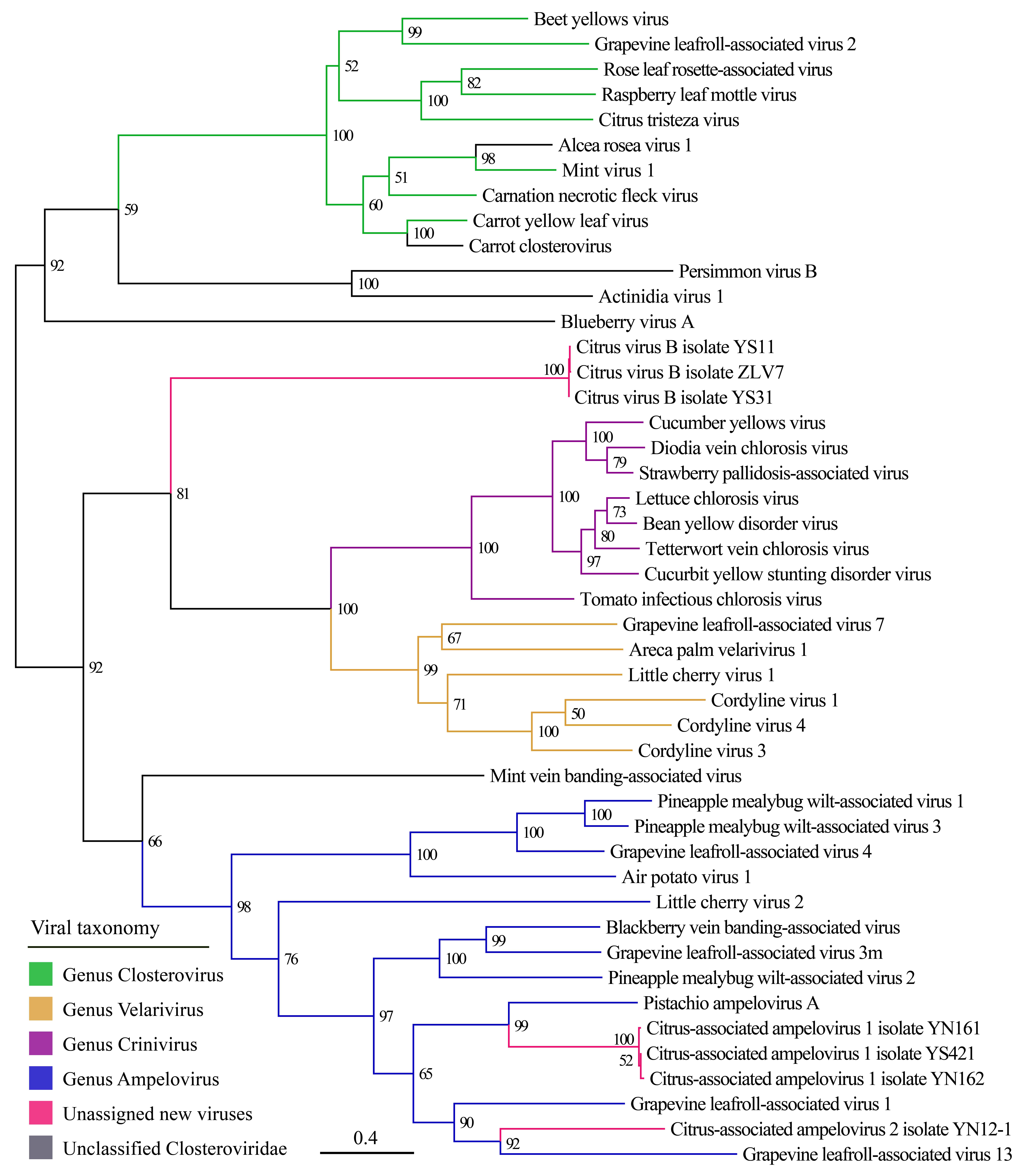
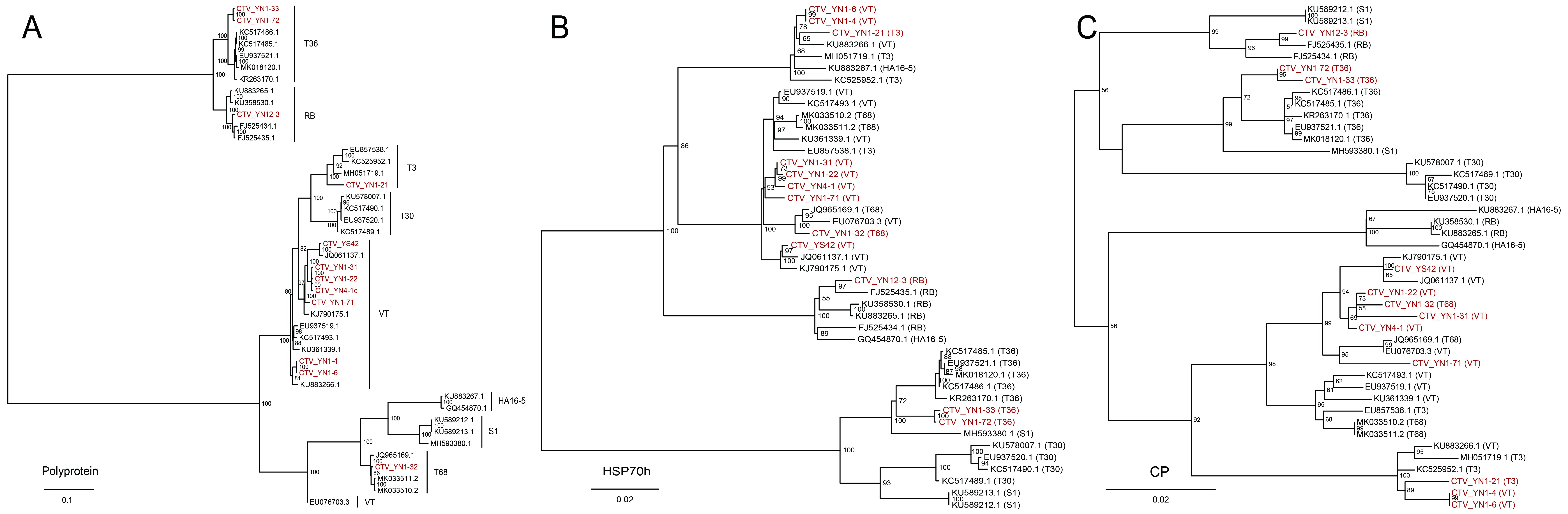
The four closterovirids that we identified and characterized from these wild hosts comprise the well-known CTV and three novel members of the family Closteroviridae: CiVB, CaAV-1, and CaAV-2. We found five genotypes of CTV that had possibly dispersed to cultivated citrus areas. Alternatively, a CTV prototype may have spread from its geographical origin to other areas via appropriate vectors, then diversified in various environments into multiple subtypes, some of which returned to the area of origin through vector(s).
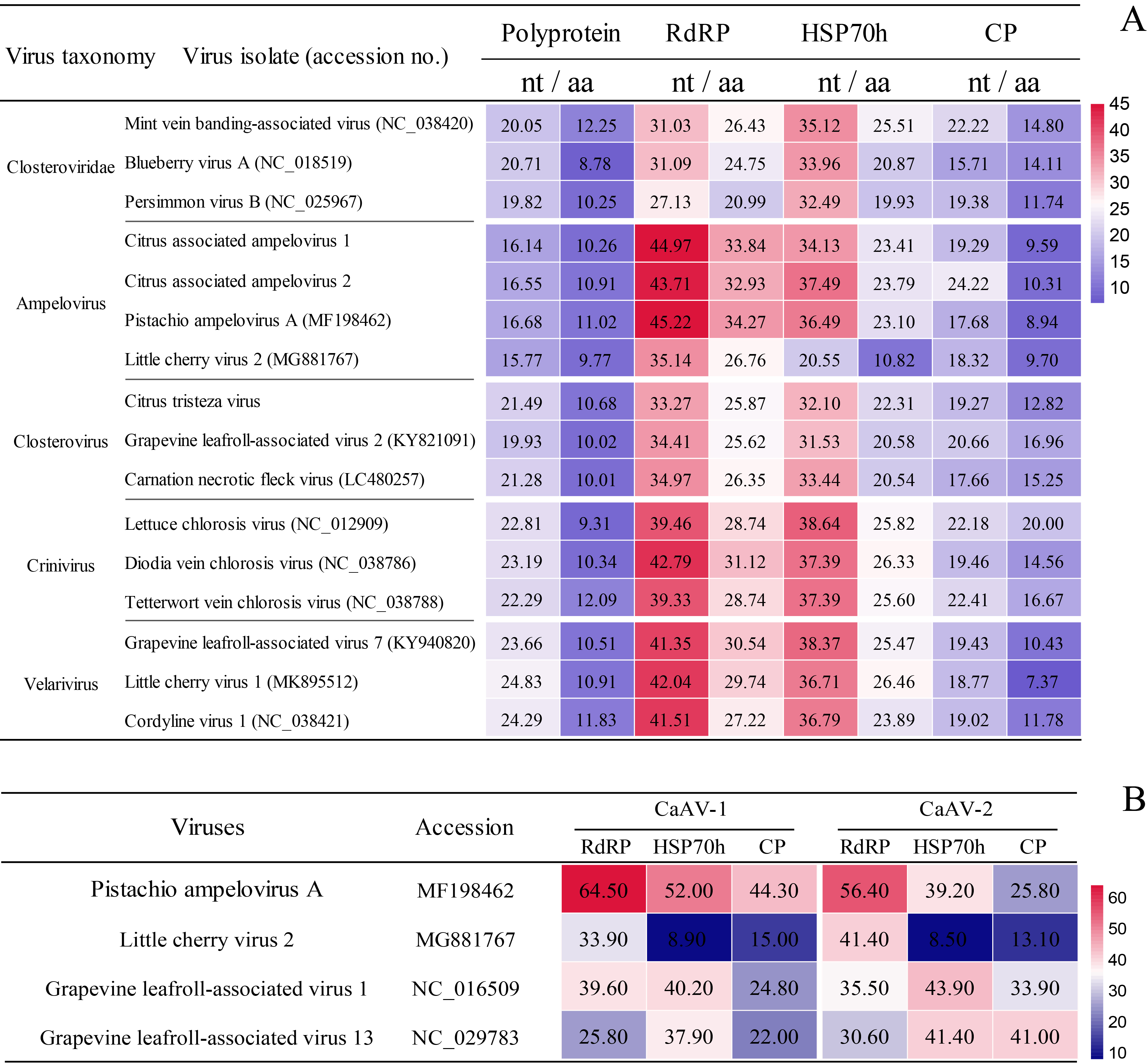
Although the prevalence and potential transmission of the novel closterovirids in other wild citrus plants is largely unknown, the present data suggest that CiVB, CaAV-1, and CaAV-2 are not as widespread as CTV. The significantly higher SNP levels in CTV than in CaAV-1 may also have led to higher evolutionary rates in CTV, a view further supported by the higher CTV intraspecific diversity found in wild citrus. Higher evolutionary rates and intraspecific diversity would result in quicker adaptation, contributing to the global distribution of CTV in citrus-growing areas.
Generally speaking, the viruses newly identified in this study show that the diversity of the family Closteroviridae exceeds what was previously thought, since the viruses reported here: i) are highly divergent compared to known members of the family, ii) have an extraordinary ability for recombination with different sources and, iii) vary widely in their genome organization and expression strategy. Recombination should affect the inferred phylogeny of CiVB, leading to the inconsistencies seen here. Our effort to determine putative inter- and intraspecific recombination events of CiVB with the RDP4 program failed to identify any reliable recombination signal. Given that other members of the family Closteroviridae may remain to be discovered, recombination events should not have been undetectable, or CiVB might be an ancestral-type virus like MVBaV [9]. In contrast to the phylogeny of MVBaV showing a closer relationship to ampeloviruses in the HSP70h and CP trees [4, 22], the HSP70h and CP phylogenies for CiVB revealed closer proximity to the bipartite criniviruses. Since CiVB is monopartite, it represents an evolutionary bridge between mono- and multipartite closterovirids. Furthermore, the characteristics of CiVB show the modular mode through which evolution operates in the family Closteroviridae (and perhaps in all RNA viruses): different genes evolving independently but within limits imposing conserved gene combinations and orders.
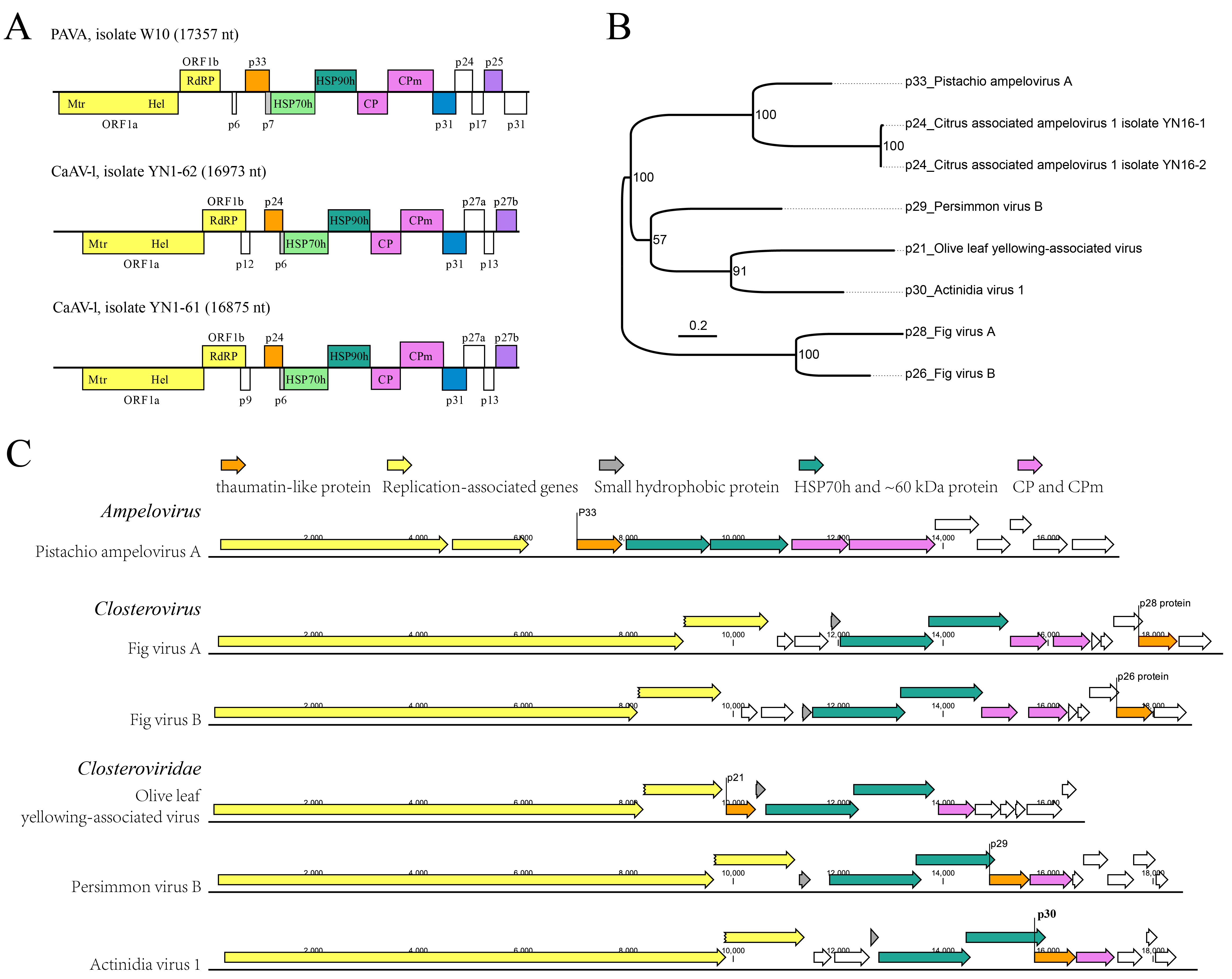
Notably, the thaumatin-like protein, which exists widely in fungi, plants, and animals and is related to host defense and developmental processes, has now been identified in seven closterovirids and is closest to the plant-derived thaumatin-like protein. The protein was only reported in these closterovirids, and it has a random genome position, unlike HSP70h that exists in all closterovirids and has a fixed genome position. However, both are encoded only by those closterovirids and not by any other viruses. Except for the thaumatin-like protein, CaAV-1 shares another two external genes with PAVA encoding two proteins containing a domain of the FieF superfamily, and a DEDDy 3'-5' exonuclease domain that have not been identified in other closterovirids.
It appears that the closterovirids have had a superior capacity to integrate exogenous genes during the long virus-plant co-evolutionary history. The members of the family Closteroviridae might share a common ancestor in which extra genes were integrated and subsequently underwent codivergence and cross-species transmission events. Whereby some genes like HSP70h have been subsequently retained in all extant closterovirids. Then, some genes have been under negative selection and are likely to remain in few species, such as thaumatin-like protein, L-Pro domains, and other unidentified proteins. Furthermore, some viruses may have been under similar stresses and thereby driven to convergent evolution via horizontally acquiring the same external genes, like CaAV-1 and PAVA. The other genes with no detectable similarity in closterovirids may diverge independently, facilitating infection of different hosts or transmission by different insect vectors.
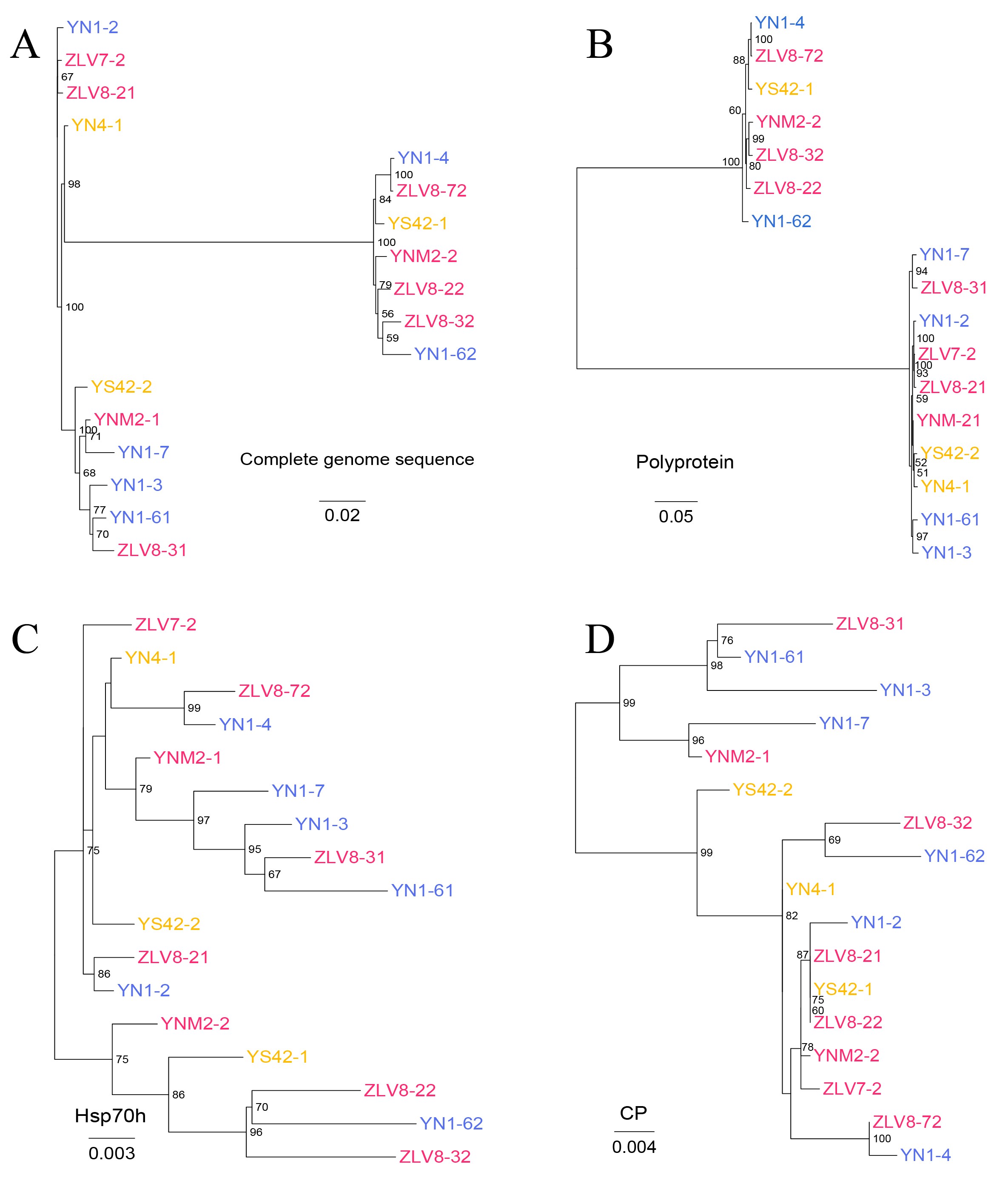
In all CTV and CaAV-1 genomes sequenced to date, the 3’-half portion of the genome is well conserved, in contrast with the 5’-half counterpart. Large fragment recombinations in the 3’-half portion of the genome are prevalent in CaAV-1. The recombination frequency in CaAV-1 showed an inverse correlation with the level of asymmetric divergence in its genome, while such a correlation was not significant in CTV. However, new CTV phylogeny lineages generated by recombination, especially of the large fragment in the 3’-half moiety of the genome, have emerged in the long history of CTV evolution [24–26]. Moreover, the tree incongruities were only present in the 3’-half portions of the CaAV-1 genomes. That indicates using a phylogenetic tree generated from complete genome sequences, or HSP70h, or CP of CTV and CaAV-1 for genotyping is not accurate. Because the translation pattern is conserved in the family Closteroviridae, CaAV-1 is likely to express a series of 3’-coterminal genes via sgRNAs, as in CTV. Thus, CTV and CaAV-1 might have used these sgRNAs for template exchanges for recombination.
The genes mapping at the 3’-half of the genome are responsible for a variety of functions important to their survival and reproduction. Although the genus Citrus comprises diverse species [10], CTV can replicate in any and all, but the viral dissemination depends on the citrus hosts [27, 28]. Sequence changes in the 3’-half portion of the genome might therefore disturb these functions and others, including vector transmission in different host species. Under selection pressure, recombination and single nucleotide variation may lead viruses to retain the more efficient fragment and to convergence of the 3’-half genome moiety in different citrus hosts.
Each wild host citrus tree was almost always coinfected with different genotypes of CaAV-1. Such a pattern might be a consequence of superinfection exclusion (SIE) of CaAV-1 not functioning, since its different genotypes showed relatively large variation. The case is similar to what happens in different CTV genotypes coinfecting citrus plants, in which primary CTV infection failing to affect secondary infection of heterologous genotypes has been reported [29]. Coinfection of one host plant by different virus genotypes might result from the same mechanism, since recombination between different genotypes is advantageous for evolution and adaptation of the virus population.
Why are there so many closterovirids in wild citrus species but not other common viruses? This may be because while CTV and perhaps the ancestor of other closterovirids are native to citrus plants, other viruses may have been transferred to cultivated citrus crops from other plants, an idea that should be further explored by examining wild citrus growing in the same areas as cultivated varieties. However, extant members of the family Closteroviridae are known to be transmitted in cultivated areas by a few specific insect vectors, whereas in a natural environment the spectrum of such vectors may be wider. Thus, in natural citrus habitats that have had no human intervention, the insect-borne closterovirids may infect wild citrus plants over a relatively wider area compared with other viruses. In addition, the Ailao Mountains are to some extent isolated from the rest of the world, and perhaps other citrus viruses and viroids that originated in other areas have not arrived here.
Apart from CTV, the discovery of CiVB, CaAV-1, and CaAV-2 represents the first report of new members of the family Closteroviridae infecting citrus plants, suggesting that these plants are potential hosts for diverse as-yet-unknown closterovirids. From one isolated region of wild citrus, we found viruses that belong to the known genus Ampelovirus (CaAV-1 and CaAV-2, two putative new species), Closterovirus (CTV), and CiVB, which may represent a new genus in the family Closteroviridae. These findings should serve as a warning that new citrus-infecting closterovirids may exist and may pose new problems for citrus production. Further surveys and monitoring are warranted to advance our knowledge and protect the citrus industry.
Viruses are a two-edged sword, affecting agriculture and natural biodiversity as they have coevolved with plants and have exerted selection pressure on their hosts. From a beneficial perspective, a few closterovirus-derived genes and RNA interference vectors have been developed to control plant diseases [30, 31]. In particular, CTV may serve as a tool for citrus tree protection and improvement [11, 32]. With a large genome similar to that of CTV, the novel citrus viruses may prove to be suitable for construction of new vectors.
The new insights acquired regarding virus ecology in wild citrus trees and genetic diversity of the family Closteroviridae support the notion that despite their genetic variation, CTV and the novel citrus closterovirids reported here evolved from the same genetic pool and that they share an ancestral-type virus, CiVB. Present-day monocultures over large areas are particularly vulnerable to a wide range of viruses, while globalization has facilitated their transport to new areas. It is in this framework, where wild and cultivated citrus species have come into contact via humans and insect vectors, that CTV became prevalent and destructive for the citrus industry. The novel viruses might disperse, as did CTV, from citrus orchards near the Ailao Mountain region. Accordingly, we need to continue monitoring and studying these novel viruses and to examine other areas harboring wild citrus in order to provide a foundation for deploying protective strategies in the future. The lessons from the recent emergence of the covid-19 pandemic apparently following a similar scenario of virus spreading from ecological niches hosting virus species to populations that were never previously exposed need to be paid attention by those concerned with citrus tree health.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}