Patient selection for placenta collection (Inclusive and Exclusive Criteria)
Patients (n=10) were selected for obtaining samples by using the following inclusion and exclusion criteria.
Inclusion criteria:
- Any age
- Any gravida
- Third-trimester Pregnancy
- Any mode of delivery
- Multifetal pregnancy
- Placental calcification grade 1 and 2
Exclusion criteria:
- 1st and 2nd-trimester pregnancies
- Placental calcification grade 3
- Torn placenta
- Any medical disorders in pregnancy
- Any placental disorder
- Abruption placentae
- Any bacterial, viral, fungal infection or infestation
- Granulomatous or necrotic patches in the placenta
- Any congenital anomaly of the fetus
- Adherent placenta (Accreta, Increta, and Percreta)
- Placental tumors
- Gestational Trophoblastic Diseases (Benign and Malignant)
Protocol for placenta collection
Vaginal Delivery: After the 2nd stage of labor, the cord was cut nearer to the neonate, keeping more than 10 cm in length of the cord attached to the placenta, and the placental end of the cut cord was clamped with artery forceps. The 3rd stage of labor was observed passively for signs of separation of the placenta. Once it was confirmed that the placenta had been separated, it was extracted using a controlled cord traction method. Slowly, with rotatory movements, the intact placenta and the membranes were obtained.
Low segment Caesarean section (LSCS): Similarly, after LSCS delivery of the fetus, the cord was cut, and the placenta was removed using controlled cord traction. Manual removal of the placenta was avoided to avoid injury to the placenta.
Grouping of the samples
The placenta samples (n=10) were grouped such as non-processed or control (Group I), decellularised (Group II), decellularized and cross-linked (Group III), re-endothelialized Group II-A, Group III-A, re-cellularized ADMSCs Group II B, and Group IIIB, co-cultured endothelial and ADMSCs Group IIC and IIIC, cultured endothelial cells Group A and ADMSCs Group B.
Placenta perfusion and decellularization
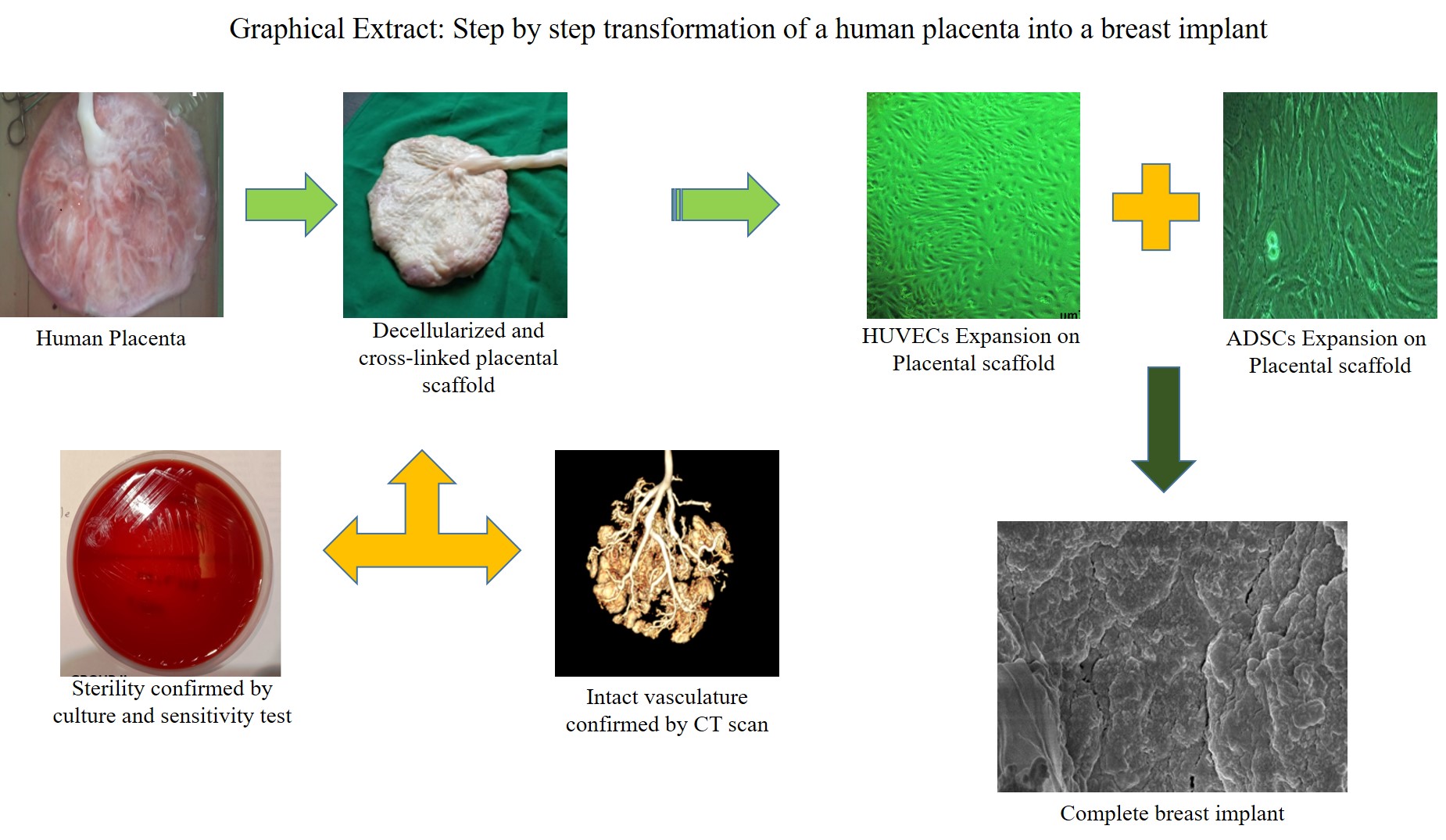
Placenta, thus obtained, was placed in a mixed solution (2000 ml) of EDTA 1% and PBS (1:1) containing antibiotics (ciprofloxacin 331.346 g/mol) after the delivery procedure. It was immediately transported to the lab for the de-cellularization process. Amniotic membranes were removed, and placenta diameter, thickness, and weight were measured and noted. The placenta was perfused with sterile distilled water and PBS 1 x till all the blood was drained (5000 ml x 3 times). Further, these cleaned placentae were subjected to a perfusion-based de-cellularization system under the influence of gravity. The onset of first step in perfusion was with 1% EDTA and PBS 1 x (1:1) ratio along with antibiotics (5000 ml) (metronidazole 5mg/ml) for 2 h. Then, it was perfused with RBC lysis buffer (5000 ml) containing antibiotic for two h. It was followed by 5000 ml of 1% EDTA and antibiotic mixture (ciprofloxacin 331.346 g/mol and metronidazole 5 mg/ml) for 2h. Once this was achieved, the placentae were subjected to distilled water mixed with an antibiotic mixture (5000 ml) (ciprofloxacin 331.346 g/mol and metronidazole 5mg/ml) for 4 h followed by 1% SDS (5000 ml) mixed with antibiotics mixture for 4h till the placenta becomes completely decellularized and translucent white. Usually, this whole process takes almost 72 hrs. The placenta was subjected to Triton-X 100 (5000ml) for 4 h and then washed with PBS 1x (3 times) (Almelkar et al. 2018, Walawalkar et al. 2020).
Cross-Linking of the decellularized placenta (Group III)
Group III (n = 10) scaffolds were cleaned by flushing their luminal surface with sterile 1× PBS (Hi-Media) and distilled water. These samples were fixed in 0.2% glutaraldehyde (GA) (SD Fine Chemicals, Mumbai, India) for 9 days and stored at 8°C. On the tenth day, these samples were treated with 10% citric acid for 10 min to neutralize the glutaraldehyde reaction (Almelkar et al. 2013). Finally, Group III cross-linked (0.2% GA) and citric acid (10%) treated were washed with sterile distilled water till neutral pH was achieved and stored at 8-degree centigrade temperature (Walawalkar and Almelkar et al. 2019).
Microbial growth assessment for processed samples
The Groups II and III scaffolds were subjected to test if any microbial growth occurs after the decellularization process by using blood agar, MacConkey’s, and Muller Hinton agar. The scaffolds pieces for both the groups were swabbed on blood agar (n=5) as well as on Maygrauled agar (n=5) surfaces (Petri dishes) using a sterile glass rod. Then the plates were incubated for 24 and 48h to observe the growth of micro-organisms or colonies or fungus.
Antibiotics sensitivity and resistance
Further, various antibiotics (total of 33) were tested to check the microbial growth sensitivity and resistance (Table 1). An antibiotic disc of 18mm diameter was used to get a zone of inhibition (microbial growth restriction) and tested the microbial sensitivity and resistance.
Scaffold treatment with antibiotics and culture
We further selected amikacin, gentamycin antibiotics based on culture sensitivity tests and perfused them through Group II and Group III scaffolds. Additionally, after 72h, re-swabbing on the blood agar, MacConkey’s, and Muller Hinton agar plates was done to check if any microbial growth occurrence persists.
CT Scan for vascular network
Radiological dye was perfused from the umbilical cord (vein and arteries) end, and it was allowed to flow through the entire placental vasculature till the dye gets out through the placental maternal end cotyledons. The placental microvasculature network CT scan was done on a CT machine, and scanned sliced imaging was done at different time points to generate the 3D placental vasculature. This process was done for Group I, II, and III placentae (maternal and fetal end) samples. Simultaneously, CT-Scan videography was generated through this process of dye perfusion. The CT-Scan sliced imaging data were analyzed using the software RadiAnt DICOM Viewer 2020.
Histology
Group I (n=10), II (n=10), and III (n=10) placental tissues were fixed in 10 % of formalin solution and further processed for dehydration and re-hydration processing. Additionally, the processed tissue samples were embedded in paraffin wax, and blocks were (fabricated) trimmed, followed by transverse section (T.S) 5 µm (Micron). The slides were sectioned for Group I, II and III and stained with Hematoxylin and Eosin (H and E) for cellular localization. The stained section was cleared in xylene solution and mounted with DPX. The slides were assessed for the nuclear and cytosolic organization on a Bright field microscope (Lynx LSM)[Walawalkar and Almelkar et al 2021; Almelkar et al 2018, Walawalkar et al 2020]. The percentage (%) of decellularization was calculated by the presence and absence of cellular nuclei using the formula.
Decellularization (%) = [Cell count for fresh Group I − Cell count decellularized Group II and Group III] × 100
Cell count for fresh Group I
Evaluation of extracellular matrix
Extracellular matrix (ECM) evaluation for was done for Group I (n=10), II (n=10), III (n=10) placental tissues. The placental tissues were fixed in 10 % of formalin solution and further processed for block sectioning with 5 µm (Micron). The slides were stained with stained with hematoxylin-eosin (HE), Verhoff – von Gieson stain (EVG), Masson Trichrome (MT), [Rieppo et al. 2019] Periodic-acid Schiff’s (PAS),[Padmanabhan et al.1985] Alician Blue 2.5 (AB 2.5) [Treesh et al. 2015].
Human umbilical vein endothelial cell isolation and culture
The isolation protocol for endothelial cells (ECs) was adopted [16, 17]. One end of the HSV was clamped using artery forceps, and 0.15% collagenase type IV (Sigma Aldrich, St. Louis, MO) and Dispase II (Roche, Nutley, NJ) were injected to detach the ECs from the tunica intima. The entire length of the vein was incubated for 20 min at 37 °C. Later, the PBS medium was flushed through the HSV (Invitrogen, Carlsbad, CA), and the digest was centrifuged at 1000 rpm for 10 min. The cell pellet was re-suspended in 10% endothelial cell growth medium-2 (ECGM-2) (Promo-Cell GmbH, Germany), and HSVECs were plated in culture flasks. At the end of 48h, ECs were fed with ECGM-2 containing 20% Fetal Bovine Serum (FBS) and 2mM L- glutamine. The HSVEC was maintained until 5 passages (Group A) [Walawalkar et al. 2020].
Cell characterization using a molecular marker (vWF)
The HUVECs (Group A) from the third subculture were used for the characterization studies. For immunocytochemistry, studies HUVECs were expanded on coverslips and were fixed using 4% paraformaldehyde. Fixed HUVECs were permeabilized using 50% methanol for 5 min followed by treatment with 5% bovine serum albumin (BSA) in PBS for 1 h to block nonspecific binding sites. Anti-von-Willebrand factor (vWF), a primary non-labeled antibody IgG fraction of anti-serum developed in the rabbit (1:200 dilution) (Chemicon, Temecula, CA), was targeted towards the HUVECs, for 12 h at 4°C, followed by respective FITC-tagged anti-rabbit secondary antibody (Invitrogen) for 1 h at 37°C. The coverslips were mounted on a mounting medium containing anti-fade (Vectashield, Vector Laboratory, Burlingame, CA) (Almelkar 2013 OA Mol Bio). The slides were then viewed using a confocal laser-scanning microscope—LSM 510 Zeiss workstation (Carl Zeiss Meditec AG, Jena, Germany) for cells in culture for vWF-/- vWF+/+. However, Group IIA (n=5) and Group IIIA (n=5) were directly signaled for expression of vWF-/- and vWF+/+ [Almelkar et al. 2013].
Isolation of breast (adipose) fat cells and their culture
A breast fat (adipose) tissue sample was obtained after the mastectomy. The sample was cleaned in PBS (1x) thoroughly to remove traces of blood clots. The washed sample was minced into small pieces and added to a 50 ml centrifuge tube with making the volume up to 50 ml with PBS. Centrifugation was done at 2000 rpm for 10 minutes, and the supernatant containing target ADSCs (lipid phase) was aspirated. The lipid phase was yellow and washed with PBS (1x) at least 2 times with the same centrifugation speed. Finally, the lipid phase, which contained the target cells were diluted with an equal volume of collagenase (0.1%) solution (Serva # 17465.02) at 37 °C in a water bath for about 30 minutes. After enzymatic digestion and incubation, an equal amount of complete medium was added and centrifuged at 2000 rpm for 10 minutes. Supernatant was discarded and cells pellets were re-suspended in NH expansion medium. The resuspended cell soup was passed through a 100µm cell filter in a separate centrifuge tube. Further, centrifugation was done at 2000rpm for 10 minutes and supernatant was discarded and fresh NH expansion medium was added. The cells were plated in 60 mm petri-dish and expanded in culture for 12 days (Group B).
Breast fat (ADSC) cell labeling (characterization) and sorting using MACS (culture)
The confluent (12 days) breast fat tissue cells were spitted, and approximately 1x 107 cells/ml were subjected to cellular characterization (MACS) and culture. Re-suspend the cell pellet in 80µl of buffer /107 cells andadd 10 μL of FcR Blocking Reagent + 10 μL of CD271-APC 10⁷ cells (Mixed well and incubate for 10mins(80C). Refresh the cells by adding 1 mL of buffer per 10⁷ cells (centrifuge at 2000rpm for 10 minutes). Aspirate supernatant and discard with the addition of 70 μL of buffer/107 cells.Add 10 μL of FcR Blocking Reagent + 20 μL of Anti-APC Micro-Beads per 10⁷ cells and incubate the sample for 15mins (80C). Wash cells by adding 1 mL of buffer / 10⁷ cells and centrifuge at 2000 rpm for 10 minutes. Aspirate supernatant entirely and re-suspend cell pellet in 500 μL of buffer per 10⁷ cells. Proceed for MACS separation by selecting the MACS column and MACS separator according to the cell density (LS column-1x108 cells). The column was placed in the magnetic field using a suitable MACS Separator. The column was rinsed with 3 mL of buffer, and the cell suspension was placed onto the column. Then unlabelled cells were passed through the column 3 times (3ml of buffer). Collect the total effluent which is unlabelled cell fraction (CD271-/-). Finally, take a new tube and remove the column from the magnetic field and place it for suitable collection. Pipette 5 mL of buffer onto the column and immediately flush out the magnetically labeled cells (CD271+/+) by firmly pushing the plunger into the column. Further proliferate the CD271+/+ cells till they become confluent (Group B).
Differentiation of ADSCs in culture.
To confirm that CD271+/+ (ADSCs) cells (Group B) have stem cell differentiation lineage properties, we subjected them to a specific differentiating signaling factor medium. We differentiated CD271+/+ (ADSCs) cells to osteogenic, adipogenic lineages. For, CD271+/+ (ADSCs) cells were cultured and expanded in a 24-well plate with DMEM with 20% FBS at a cell density of 1x 10 3 cells /ml. Further, differentiation to adipose lineage was achieved in the presence of dexamethasone-4mg/ml (1:1000), Insulin- 40 mg/ml (1:200) and isobutylmethylxanthine (0.5mM). The cells were expanded and maintained with a conditioning medium for about 15 to 18 days. Oil Red O staining was done to study fat deposition in plasticity remodeled cells to adipogenic cell phenotype after fixation with 4% paraformaldehyde. However, for CD271+/+ (ADSCs) cells differentiation to osteogenic lineages, the cells were treated with 10 mM beta-glycero-phosphate (Merck, Darmstadt, Germany), ascorbic acid 2-phosphate (50 g/ml) and Dexamethasone (4 mg /ml- 1: 1000) (Sigma). The cells were expanded and maintained with a conditioning medium for about 15 days. The generation of calcium phosphate deposits was assessed by staining with Alizarin Red S (Sigma. USA) for 10 min at 4ºC, after fixing the cells with 4 % paraformaldehyde.
Field emission scanning electron microscopy (FE-SEM)
FE-SEM of Group I, Group II, Group III, Group IIA, Group IIIA, Group IIB, Group IIIB, Group IIC, and Group IIIC was performed. HUVECs expansion onto the fibrin glue (FG) coated surface was analyzed for endothelization and CD271+/+ (ADSCs) expansion (Almelkar OA Tissue 2013). All group samples were fixed in 2.5% glutaraldehyde for 2h at room temperature. Later, they washed with PBS 1x and dehydrated with increasingly gradient ethanol for 10 min each. Further, samples were dried overnight and sputter-coated with gold (Quorum Technologies Ltd., UK) and observed under FE-SEM (Nano NOVA SEM NPEP303)[Walawalkar and Almelkar et al. 2019; Eswaramoorthy et al. 2018].
Recellularization and cell expansion.
Sterilized and antibiotic-treated decellularised Group II (n=10) and III (n=10) scaffolds were seeded with 500 µl of HUVECs, CD271+/+ ADSCs and co-culture of HUVECs +CD271+/+ ADSCs suspensions with a cell density of 10 × 104 cells/ml. Subsequent incubation at 37 °C in a humidified atmosphere for 5 hours was performed to encourage cells to diffuse into and adhere to the scaffold. The cell-loaded scaffolds were termed Group IIA (n=10), Group IIB (n=10), Group IIC (n=10) and Group IIIA (n=10), Group IIIB (n=10) and Group IIIC (n=10) which was then further incubated in ECGM-2 containing 10% FBS for HUVECs, and the (DMEM) culture media (ADSCs) was refreshed every 5−6 days. Simultaneously HUVECs (Group A, n=10) and CD271+/+ ADSCs cells (Group B, n=10) were cultured in Petri plates separately to understand their cell density after reaching confluence in culture.
Cell viability and non-viability assay
3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (Himedia, Mumbai, India) (5μg/ml) was used for cell-based biochemical assay for confirmation of HUVEC expansion on the surface of Group IIA (n=10) and IIIA (n=10) scaffolds (10,000 cells/II and III scaffold) and tissue culture plate (10,000 cells/well), CD271+/+ (ADSCs) expansion on the surface of Group IIB (n=10) and Group IIIB (n=10), co-culture of HUVECs+ CD271+/+ (ADSCs) expansion was done Group IIC (n=10) and Group IIIC (n=10), HUVECS in culture were Group A (n=10) and CD271+/+ (ADSCs) expansion were Group B (n=10). MTT assay was performed at various day points such as 0 day, 4day, 8 days, and 12 days. HUVECs and CD271+/+ (ADSCs) expansion expanded by approximately 2 cm in length in all groups scaffold [Walawalkar et al 2020].
Nitric oxide levels on cells in culture and scaffolds.
Nitric oxide (NO) assay (Himedia, India) was done for the quantification and its release from Group I (n=10), Group II (n=10), Group III (n=10), Group IIA (n=10), Group IIIA (n=10), Group IIB (n=10), Group IIIB (n=10), Group IIC (n=10)and Group IIIC (n=10), Group A (n=10) and Group B (n=10). The samples were crushed, and 200 µl of the gradient of each sample was aliquoted in Eppendorf tubes. Approximately 10 µl of reducing agent (R1) and 10 µl of Griess Reagent 1 & 2 (GR 1 & 2) were added, and the samples were incubated in a dark room at room temperature for 3 h. The absorbance was taken at 530 nm, and total NO levels were quantified using monochromator absorbance (Synergy Biotek: SFLX).
Biochemical peroxidation and antioxidant levels for cultured and scaffold expanded cells
Oxidative stress (BIOXYTECH LPO-586, US) using Malondialdehyde (MDA) levels were quantified for Group I, Group II, Group III, Group IIA, Group IIIA, Group IIB, Group IIIB, Group IIC, Group IIIC and Group A and Group B. All samples were crushed in chilled lysis buffer [Tris buffer (20mM, pH 7.4)]. Approximately 200 µl of suspension for the assay and added 650 µl R1 reagent (N-methyl-2-phenylindole in acetonitrile) and 150 µl of 1N HCl to the reaction mixture. The reaction mixtures were incubated for 1 h at 800C. MDA (peroxidation) levels were quantified at absorbance 520nm using monochromator absorbance (Synergy Biotek: SFLX) [Walawalkar and Almelkar et al. 2019].
The antioxidant (catalase) levels were estimated for expanded (Group IIA, IIB, IIC, A, B, and Group IIIA, IIIB, IIIC, A, B) and non-expanded (Group II, Group III) HUVECs. Group I was served as a control sample. Simultaneously the catalase activity was also quantified for HUVECs in culture. We used the EnzyChromTM Catalase assay kit (ECAT-100) for the quantitative colorimetric estimation of the catalase levels. We dissolved all samples in 400 µl of assay buffer (3% H2O2), from which 102 µl of the mixture was utilized for the quantification. 1 µl of dye reagent and 1 µl HRP enzyme were added to this mixture, incubated for 30 mins, and the absorbance was obtained on 570 nm using monochromator absorbance (Synergy Biotek: SFLX)[Walawalkar et al. 2020].
Picogreen Assay
The samples in groups I, II, III, IIA, IIIA, IIB, IIIB, IIC, III C, A, and B (n = 10) were placed in lysis buffer for 10 min and macerated. 40 μL of each sample was obtained in a 5 ml cuvette, and 3.96 mL of distilled water was added, followed by DNA samples incubation for 15 min. The absorbance was measured at 260 nm with ds-DNA levels quantified by a PicoGreen fluorescent assay to confirm the occurrence of residual tissue ds-DNA in the decellularized group. All samples were compared to group I. However, for cellular expansion (DNA duplication) on placental scaffolds (biocompatibility) and re-endothelization and re-cellularization, co-cultures of scaffolds were also quantified by PicoGreen fluorescence assay.
Statistical analysis
Graphpad Prism 5 software was used to obtain mean ± SD, and a one-way ANOVA (***p < 0.05) test was performed, and the graph was also obtained. Graph Pad Prism 5 software was used to perform a one-way analysis of variance (ANOVA, ***p<0.0001) including mean and standard deviation (mean ± SD). Dunnett’s test was used to compare control groups (mean ± SD) with all other decellularized groups (mean ± SD).
{kind=link}