General Experimental Methods and Instruments
All solvents and reagents, unless otherwise noted, were purchased from commercial sources and used as received without further purification. Solvents noted as ‘’dry’’ were obtained following storage over 3 Å molecular sieves. The bifunctional ligand 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid mono-N-hydroxysuccinimide ester (DOTA-NHS-ester) was purchased from Macrocyclics (Plano, TX). Trans-cyclooctene-N-hydroxysuccinimide (TCO-NHS) was purchased from Click Chemistry Tools (Scottsdale, Az). Nuclear Magnetic Resonance (NMR) spectra were recorded using a 400, 500 & 600 MHz spectrometer. Chemical shifts are reported relative to internal Me4Si in CDCl3 (δ 0.0) or CDCl3 (δ 7.26), (CD3)2SO (δ 2.50), CD3CN (δ 1.94) HOD for D2O (δ 4.65) and CD2HOD (d 3.31) for 1H and CDCl3 (δ 77.16), (CD3)2SO (δ 39.5), CD3CN (δ 118.26) and CD3OD (d 49.0) for 13C. 1H-NMR signals were assigned with the aid of COSY. 13C signals were assigned with the aid of DEPT-135, HSQC and HMBC. Coupling constants (J) are reported in Hertz and are reported uncorrected. High resolution mass spectra (HRMS) were measured in positive and/or negative mode as indicated using CH3CN, H2O and/or MeOH as solvent using an Agilent LC Mass Spectrometry instrument. Infrared (IR) spectra were recorded neat on a Perkin Elmer Spectrum Two FTIR spectrometer. Only selected, characteristic absorption data are provided for each compound. Reactions were monitored by thin layer chromatography (TLC), performed on aluminium sheets pre-coated with Silica Gel 60 (HF254, E. Merck) and spots visualized by UV and charring with cerium (IV) molybdate solution, vanillin, permanganate, anisaldehyde or ninhydrin solutions. During reaction work-ups a TLC of each extractant was taken to ensure complete retention of the product in the organic layer. Flash column chromatography was generally employed and was carried out using silica gel 60 (0.040-0.630 mm) using a stepwise solvent polarity gradient correlated with TLC mobility. Chromatography solvents used were diethyl ether, toluene, hexane, acetonitrile, EtOAc, CH2Cl2, MeOH (Sigma Aldrich). The analysis and purification of polar compounds were carried out using an Agilent 1100 HPLC and PDA detector at 254 nm with conditions: Kinetex (10 x 150 mm, 5 µm) with a gradient of acetonitrile: 0.1% TFA from 80:20 to 50:50 over 15 min with a flow rate of 2 mL/min.
Luminescence experiments were conducted on an Edinburgh Instruments FS5 spectrometer using the SC-05 cassette. All spectra were corrected for instrument response. Excitation and emission monochromator bandwidths for 7 and 17 were set to 1 nm, and 1.5 nm for 13. The emission spectrum for 13 was collected with the aid of a Knight Optical 395 nm long-pass filter to reduce the effects of scattering. Solution-based absolute photoluminescent quantum yields (PLQY) were obtained for 7, 13, and 17 via the SC-30 Integrating Sphere cassette with excitation wavelengths of 380, 490, and 378 nm respectively.
The average number of TCO moieties per antibody, or number of DO3A-BODIPY-Tz scaffolds conjugated to trastuzumab-TCO was determined by MALDI-ToF MS/MS on a Bruker autoflex speed at the Alberta Proteomics and Mass Spectrometry Facility (University of Alberta, Canada) using previously described procedures.19,44 The [M+2H]2+ mass signals from the chromatograms of purified trastuzumab and each conjugate was used to determine the average mass, and the TCO-to-protein or ligand-to-proton ratio for each was determined by subtracting the molecular weight of trastuzumab from the molecular weight of the conjugate, and then dividing by the mass of the scaffold.
Synthesis and Characterization of Compounds and Complexes
Compounds 2, 20, 21 were prepared using a modified procedure than reported.19
Fmoc-Lys(Boc)-OSu (20). N-hydroxysuccinimide (245 mg, 2.13 mmol) and EDC (408 mg. 2.13 mmol) were added to a solution of Fmoc-Lys(Boc)-OH (1 g, 2.13 mmol) in dry CH2Cl2 (42 mL). After stirring the reaction mixture for 16 h under nitrogen, the solvent was removed under reduced pressure and the resulting residue was dissolved in CH2Cl2 (20 mL). The solution was washed with water (20 mL), dried over Na2SO4, filtered and the solvent was removed under reduced pressure to give Fmoc-Lys(Boc)-OSu (1.21 g, 100%) as a white solid. The 1H NMR data for the productwas in good agreement with those previously reported in the literature19; Rf 0.24 (hexane-EtOAc 1:1); IR (film) cm-1: 3338, 2932, 2864, 1739, 1673, 1691, 1523, 757, 736, 646, 587; 1H NMR (CDCl3/MeOD (1 drop), 400 MHz) δ 7.76 (2H, d, J 7.7, Ar-H), 7.60 (2H, dt, J 7.5, 1.8, Ar-H), 7.39 (2H, td, J 7.5, 1.1, Ar-H), 7.31 (2H, td, J 7.5, 1.2, Ar-H), 4.77 – 4.66 (1H, m, FmocNHCH), 4.49 – 4.37 (2H, m, CHCH2), 4.23 (1H, t, J 7.0, CHCH2), 3.13 (2H, t, J 6.0, CH2NHBoc), 2.83 (4H, s, O=CCH2CH2C=O), 2.06 – 1.83 (2H, m, CHaHb), 1.60 – 1.47 (4H, m, CH2 x 2), 1.43 (9H, s, t-Bu); 13C NMR (CDCl3, 125 MHz) δ 168.6, 168.2, 156.2, 155.7 (each C=O), 143.6, 141.3, 127.7, 127.1, 125.1, 120.0 (each Ar-C), 79.2 (C(CH3)3), 67.3 (CHCH2), 52.2 (FmocNHCH), 47.1 (CHCH2), 39.8 (CH2NHBoc), 31.9 (CHaHb), 29.4 (CH2), 28.4 (C(CH3)3), 25.6 (O=CCH2CH2C=O), 21.9 (CH2); ESI-HRMS calcd. C30H39N4O8, 583.2762 found m/z 583.2743 [M+NH4]+.
Fmoc-Lys(Boc)-Lys(Z)-OtBu (21)19. A suspension of L-Lys(Z)-OtBu·HCl (738 mg, 1.98 mmol) in dry CH2Cl2 (8 mL) was treated with DIPEA (0.35 mL, 1.98 mmol). The resulting mixture was added to a solution of Fmoc-Lys(Boc)-OSu 20 (792 mg, 1.4 mmol) in dry CH2Cl2 (8 mL) at 0 °C. The mixture was warmed to room temperature and stirred for 16 h under argon, then washed with brine (10 mL, dried over Na2SO4, filtered and the solvent was removed under reduced pressure. Flash chromatography of the residue (hexane-EtOAc, 2:1 – 1:2) gave the title compound (1.04 g, 95%) as a white solid; Rf 0.5 (hexane-EtOAc, 1:2); IR (film) cm-1: 3311, 2932, 2864, 1683, 1651, 1531, 1249, 1160, 734, 645; 1H NMR (MeOD, 400 MHz) δ 7.80 (2H, d, J 7.5, Ar-H), 7.66 (2H, t, J 7.6, Ar-H), 7.40 (2H, t, J 7.5, Ar-H), 7.36 – 7.24 (7H, m, each Ar-H), 5.12 – 4.95 (2H, m, OCH2C6H5), 4.37 (2H, d, J 6.9, CHCH2), 4.28 (1H, dd, J 8.9, 5.0, NHCH), 4.21 (1H, t, J 6.9, CHCH2), 4.12 (1H, dd, J 8.8, 5.4, NHCH), 3.17 – 2.99 (4H, m, CH2NHBoc & CH2NHCbz), 1.90 – 1.58 (4H, m, CHaHb & CHa’Hb’), 1.57 – 1.32 (26H, m, each CH2 x 4 & t-Bu x 2); 13C NMR (MeOD, 100 MHz) δ 174.8, 172.7, 158.9, 158.5, 158.4 (each C=O), 145.3 (2s), 142.6, 138.4, 129.4, 128.9, 128.8, 128.7, 128.2 (2s), 126.2, 120.9 (each Ar-C), 82.8 (C(CH3)3), 79.9 (C(CH3)3), 67.9 (CHCH2), 67.3 (OCH2C6H5), 56.3 (NHCH), 54.3 (NHCH), 48.5 (CHCH2), 41.5, 41.1 (CH2NHBoc & CH2NHCbz), 32.9, 32.3, 30.5, 30.3 (each CH2), 28.8 (C(CH3)3), 28.3 (C(CH3)3), 24.1, 23.9 (each CH2); ESI-HRMS calcd. C44H62N5O9, 804.4542 found m/z 804.4504 [M+NH4]+..
H2N-Lys(Boc)-Lys(Z)-OtBu (2). To a stirred solution of Fmoc-Lys(Boc)-Lys(Z)-OtBu 21 (640 mg, 0.81 mmol) in dry CH2Cl2 (8 mL) was added NHEt2 (1.7 mL, 16.3 mmol). The mixture was stirred at room temperature for 5 h under argon. The solvent was removed under reduced pressure and flash chromatography of the residue (CH2Cl2-MeOH, 100:0 – 98:2 – 95:5 – 90:10) gave 2 (456 mg, 99%) as a colourless oil which spontaneously solidified over time; Rf 0.4 (CH2Cl2-MeOH 90:10); IR (film) cm-1: 3355, 2933, 2864, 1721, 1686, 1519, 1165, 1245, 723, 697, 631; 1H NMR (MeOD, 400 MHz) δ 7.42 – 7.24 (5H, m, Ar-H), 5.06 (2H, m, OCH2C6H5), 4.26 (1H, dd, J 8.5, 5.2, NHCH), 3.34 (1H, m, NH2CH), 3.12 (2H, t, J 6.8, CH2NHCbz), 3.04 (2H, t, J 6.8, CH2NHBoc), 1.88 – 1.34 (30H, m, each CH2 x 6 & t-Bu x 2); 13C NMR (MeOD, 100 MHz) δ 177.5, 172.8, 158.9, 158.5 (each C=O), 138.4, 129.4, 128.9, 128.7 (each Ar-C), 82.9 (C(CH3)3), 79.8 (C(CH3)3), 67.3 (OCH2C6H5), 55.8 (NH2CH), 54.3 (NHCH), 41.5 (CH2NHCbz), 41.1 (CH2NHBoc), 36.1 (CH2), 32.4 (CH2), 30.7 (CH2), 30.4 (CH2), 28.8 (C(CH3)3), 28.3 (C(CH3)3), 24.0 (CH2), 23.8 (CH2); ESI-HRMS calcd. C29H49N4O7, 565.3601 found m/z 565.3618 [M+H]+.
4,4-difluoro-8-(4-(succinimidocarboxy)phenyl)-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-s-indacene (1)50. N-hydroxysuccinimide (248 mg, 2.16 mmol) and EDC (414 mg. 2.16 mmol) were added to a solution of 4,4-difluoro-8-(4-carboxyphenyl)-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-s-indacene acid50 (918 mg, 2.16 mmol) in dry CH2Cl2 (45 mL). After stirring the reaction mixture for 16 h under argon, the solvent was removed under reduced pressure and the resulting residue was dissolved in CH2Cl2 (20 mL). The solution was washed with water, dried over Na2SO4, filtered and the solvent was removed under reduced pressure. Flash chromatography of the residue (CH2Cl2, 100%) gave 1 (662 mg, 59%) as a red-orange solid. The 1H and 13CNMR data for 1 was in good agreement with those previously reported in the literature50; Rf 0.24 (CH2Cl2 100%); IR (film) cm-1: 2967, 2929, 2868, 1763, 1740, 1538, 1187, 976, 727, 533; 1H NMR (CDCl3, 400 MHz) δ 8.27 (1H, d, J 8.6, Ar-H), 7.50 (1H, d, J 8.6, Ar-H), 2.96 (4H, s, O=CCH2CH2C=O), 2.54 (6H, s, CH3 x 2), 2.31 (4H, q, J 7.5, CH2CH3), 1.28 (6H, s, CH3 x 2), 0.99 (6H, t, J 7.5, CH2CH3); 13C NMR (CDCl3, 125 MHz) δ 169.2, 161.4 (each C=O), 154.6, 143.0, 138.0, 137.7, 133.3, 131.2, 130.1, 129.3, 125.6 (each Ar-C), 25.7 (O=CCH2CH2C=O), 17.0 (CH2CH3), 14.6 (CH2CH3), 12.6, 12.1 (each CH3); ESI-HRMS calcd. C28H34BF2N4O4, 539.2641 found m/z 539.2627 [M+H]+.
4,4-dicyano-8-(4-(succinimidocarboxy)phenyl)-2,6-diethyl-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-s-indacene (7). To a stirred solution of 1 (100 mg, 0.192 mmol) and BF3.OEt (0.24 mL, 1.92 mmol) in dry CH2Cl2 (20 mL) was added TMS-CN (0.48 mL, 3.84 mmol). The reaction mixture was stirred in the dark at room temperature for 1 h. EtOAc (40 mL) was added and the organic later was washed with water (20 mL), brine (20 mL), dried over Na2SO4 and the solvent was removed under reduced pressure. Flash chromatography of the residue (hexane-EtOAc, 6:4 – 1:1) gave 7 (79 mg, 77%) as a red-orange solid; Rf 0.4 (hexane – EtOAc, 1:1); IR (film) cm-1: 2956, 2923, 2854, 1768. 1740, 1540, 1474, 1185, 977, 726, 544; 1H NMR (CDCl3, 400 MHz) δ 8.31 (2H, d, J 8.3, Ar-H), 7.51 (2H, d, J 8.3, Ar-H), 2.96 (4H, s, O=CCH2CH2C=O), 2.71 (6H, s, CH3 x 2), 2.37 (4H, q, J 7.6, CH2CH3), 1.32 (6H, s, CH3 x 2), 1.02 (6H, t, J 7.6 CH2CH3); 13C NMR (CDCl3, 100 MHz, CN signals were not observed) δ 169.1, 161.2 (each C=O), 155.0, 141.8, 139.5, 138.6, 134.8, 131.4, 129.1, 128.6, 126.2 (each Ar-C), 25.7 (O=CCH2CH2C=O), 17.2 (CH2CH3), 14.4 (CH2CH3), 13.5, 12.3 (each CH3); ESI-HRMS calcd. C30H31BN5O4, 536.2469 found m/z 536.2467 [M+H]+.
tert-butyl N6-((benzyloxy)carbonyl)-N2-(N6-(tert-butoxycarbonyl)-N2-(4-(4,4-dicyano-2,6-diethyl-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-s-indacen-8-yl)benzoyl)-L-lysyl)-L-lysinate (8). A solution of the amine 2 (168 mg, 0.3 mmol) and triethylamine (41 µL, 0.3 mmol) in dry CH2Cl2 (1.5 mL) was added under argon to a solution of the succinimide 7 (79 mg, 0.15 mmol) in dry CH2Cl2 (1.5 mL). The reaction mixture was stirred in the dark at room temperature for 4 days. The solvent was removed under reduced pressure and flash chromatography of the residue (CH2Cl2-CH3CN 4:1 – 2:1) gave 8 (116 mg, 80%) as a red-orange solid; Rf 0.3 (EtOAc-hexane 1:1); IR (film) cm-1: 2969, 2978, 2863, 1699, 1643, 1541, 1188, 1153, 734, 544; 1H NMR (MeOD, 500 MHz) δ 8.08 (2H, d, J 8.3, Ar-H), 7.41 (2H, d, J 8.1, Ar-H), 7.35 – 7.19 (5H, m, OCH2C6H5), 5.07 (1H, d, J 12.6, OCHHC6H5), 5.03 (1H, d, J 12.6, OCHHC6H5), 4.59 (1H, dd, J 8.8, 5.9, NHCH), 4.32 (1H, dd, J 9.1, 4.9, NHCH), 3.17 – 3.02 (4H, m, CH2NHBoc & CH2NHCbz), 2.68 (6H, s, CH3 x 2), 2.44 (4H, q, J 7.5, CH2CH3), 1.99 – 1.81 (3H, m, CHaHb & CHa’Hb’), 1.77 – 1.66 (1H, m, CHa’Hb’), 1.62 – 1.40 (26H, m, each CH2 x 4 & t-Bu x 2), 1.37 (6H, s, CH3 x 2), 1.03 (6H, t, J 7.5, CH2CH3); 13C NMR (MeOD, 125 MHz, , CN signals were not observed) δ 174.5, 172.8, 169.3, 158.9, 158.6 (each C=O), 155.5, 142.1, 141.6, 139.2, 138.5, 136.4, 136.1, 130.2, 129.9, 129.8, 129.4, 128.9, 128.8 (each Ar-C), 82.8 (C(CH3)3), 79.9 (C(CH3)3), 67.3 (OCH2C6H5), 55.5 (NHCH), 54.4 (NHCH), 41.6, 41.1 (CH2NHBoc & CH2NHCbz), 32.7, 32.2, 30.7, 30.3 (each CH2), 28.8 (C(CH3)3), 28.3 (C(CH3)3), 24.5, 24.0 (each CH2), 17.9 (CH2CH3), 14.8 (CH2CH3), 13.5 (CH3), 12.5 (CH3); ESI-HRMS calcd. C55H74BN8O8, 985.5723 found m/z 985.5747[M+H]+.
N6-((benzyloxy)carbonyl)-N2-(4-(4,4-dicyano-2,6-diethyl-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-s-indacen-8-yl)benzoyl)-L-lysyl)-L-lysine (9). To a solution of 8 (50 mg, 51 µmol) in dry CH2Cl2 (1.6 mL) was added TFA (0.4 mL) dropwise. The reaction mixture was stirred in the dark at room temperature for 6 h. The solvent was removed under a stream of air and the resulting residue was washed with three 10 mL portions of Et2O to give the amine 9 (48 mg, 100%) as a red orange solid; 1H NMR (MeOD, 500 MHz) δ 8.09 (2H, d, J 8.1, Ar-H), 7.43 (2H, d, J 8.1, Ar-H), 7.36 – 7.21 (5H, m, OCH2C6H5), 5.09 (1H, d, J 12.5, OCHHC6H5), 5.05 (1H, d, J 12.5, OCHHC6H5), 4.65 (1H, t, J 7.2, NHCH), 4.48 (1H, dd, J 9.5, 4.4, NHCH), 3.19 – 3.07 (2H, m, CH2), 2.99 (2H, t, J 7.6, CH2), 2.70 (6H, s, CH3 x 2), 2.46 (4H, q, J 7.5, CH2CH3), 2.05 – 1.87 (3H, m, CH2 & CH), 1.82 – 1.73 (3H, m, CH2 & CH), 1.65 – 1.47 (6H, m, each CH2), 1.39 (6H, s, CH3 x 2), 1.05 (6H, d, J 7.5, CH2CH3); 13C NMR (MeOD, 125, CN signals were not observed) δ 175.3, 174.2, 169.3, 158.9 (each C=O), 155.5, 141.8, 141.6, 139.4, 138.6, 136.3, 136.1, 130.1, 129.9, 129.8, 129.4, 128.9, 128.7 (each Ar-C), 67.3 (OCH2C6H5), 55.2 (NHCH), 53.5 (NHCH), 41.6, 40.6 (CH2NH2 & CH2NHCbz), 32.3, 32.2, 30.3, 28.2, 24.1, 23.8 (each CH2), 17.9 (CH2CH3), 14.8 (CH2CH3), 13.5 (CH3), 12.5 (CH3); ESI-HRMS calcd. C46H58BN8O6, 829.4572 found m/z 829.4554 [M+H]+.
N6-((benzyloxy)carbonyl)-N2-(N6-(2-(4-(1,2,4,5-tetrazin-3-yl)phenyl)acetyl)-N2-(4-(4,4-dicyano-2,6-diethyl-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-s-indacen-8-yl)benzoyl)-L-lysyl)-L-lysine (11). After drying under high vacuum for 2 h the amine 9 (48 mg, 51 µmol) was dissolved in dry CH2Cl2 (4 mL) and DIPEA (88 µL, 510 µmol) was added. Tetrazine 10 (19 mg, 61.2 µmol) was added in one portion and the reaction mixture was stirred in the dark at room temperature for 2 h. The mixture was diluted with CH2Cl2 (30 mL), washed with 0.5 M HCl (10 mL), water (10 mL), brine (10 mL), dried over Na2SO4, filteredand the solvent was removed under reduced pressure. Flash chromatography of the residue (100% CH2Cl2 – CH2Cl2-MeOH 95:5 – 80:20) gave 11 (47 mg, 90% for two steps) as a red solid; Rf 0.54 (CH2Cl2-MeOH, 9:1); IR (film) cm-1: 3313. 2931, 1547, 1190, 1155, 981, 736; 1H NMR (MeOD, 500 MHz) δ 10.29 (1H, s, Tetrazine-H), 8.53 (2H, d, J 8.3, Ar-H), 8.07 (2H, d, J 8.1, Ar-H), 7.57 (2H, d, J 8.3, Ar-H), 7.39 (2H, d, J 8.1, Ar-H), 7.33 – 7.17 (5H, m, each Ar-H), 5.06 (1H, d, J 12.6, OCHHC6H5), 5.02 (1H, d, J 12.6, OCHHC6H5), 4.59 – 4.55 (1H, m, NHCH), 4.40 – 4.34 (1H, m, NHCH), 3.65 (2H, s, CH2), 3.28 – 3.24 (2H, m), 3.14 – 3.06 (2H, m), 2.68 (6H, s, CH3 x 2), 2.42 (4H, d, J 7.6, CH2CH3), 2.00 – 1.82 (3H, m, CHaHb & CHa’Hb’), 1.79 – 1.70 (1H, m, CHa’Hb’ ), 1.66 – 1.41 (6H, m, each CH2), 1.34 (6H, s, CH3 x 2), 1.02 (6H, t, J 7.6, CH2CH3 ); 13C NMR (125 MHz, MeOD, CN signals were not observed) δ 174.0 (2s), 173.1, 168.8 (each C=O), 167.5 (Tetrazine-C), 161.9 (Tetrazine-C), 158.8 (C=O), 155.4, 143.4, 142.0, 141.4, 139.8, 138.4, 137.2, 136.1, 131.9, 131.2, 130.1, 129.9, 129.8, 129.7, 129.4, 129.3, 128.9, 128.7 (each Ar-C), 67.3 (OCH2C6H5), 55.5 (NHCH), 52.6 (NHCH), 43.8 (CH2C8H5N4), 41.7 (CH2NHCbz), 39.9 (CH2NHCO), 32.4 (2s), 30.3, 29.7, 24.4, 24.1 (each CH2), 17.9 (CH2CH3), 14.8 (CH2CH3), 13.6 (CH3), 12.6 (CH3); ESI-HRMS calcd. C56H62BN12O7, 1025.4957 found m/z 1025.4982 [M-H]-.
N2-(N6-(2-(4-(1,2,4,5-tetrazin-3-yl)phenyl)acetyl)-N2-(4-(4,4-dicyano-2,6-diethyl-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-s-indacen-8-yl)benzoyl)-L-lysyl)-L-lysine (12). To a stirred solution of 11 (80 mg, 77.9 µmol) in EtOH (44 mL) and formic acid (88 µL) was added 10% palladium on carbon (144 mg). The reaction flask was stoppered and flushed with argon for 1 min followed by hydrogen for a further 1 min. The mixture was stirred at room temperature in the dark under hydrogen (balloon) for 6 h, filtered through Celite and the solvent was removed under reduced pressure. The residue was washed with three 10 mL portions of each Et2O, EtOAc and hexane to give the formate salt of 12 (52.5 mg, 72%) as a red solid which was taken to the next step without further purification. A portion of the crude residue was purified via C-18 column (H2O (0.1% TFA) – ACN (0.1% TFA) 35:75 – 45:55) to give the TFA salt of 12 for characterisation. Important note: prior to obtaining NMR data of the crude formate salt, the solution of the compound in MeOD is heated to 40 °C for 15 minutes to ensure complete conversion of exchangeable hydrogens with deuterium. The exchange was found to be slow at room temperature and, if not given the appropriate time to exchange, can result in an artifact where multiple compounds are present in the NMR simultaneously; 1H NMR (MeOD, formate salt, 500 MHz) δ 10.31 (1H, s, Tetrazine-H), 8.56 (2H, d, J 8.3, Ar-H), 8.52 (1H, s, HCOOH), 8.11 (2H, d, J 8.3, Ar-H), 7.60 (2H, d, J 8.3, Ar-H), 7.53 (2H, d, J 8.3, Ar-H), 4.53 (1H, dd, J 8.9, 5.6, NHCH), 4.33 (1H, dd, J 7.7, 4.9, NHCH), 3.68 (2H, s, CH2), 3.29 (2H, t, J 6.6, CH2), 2.95 (2H, t, J 7.4, CH2), 2.70 (6H, s, CH3 x 2), 2.45 (4H, q, J 7.6, CH2CH3), 2.00 – 1.50 (12H, m, each CH2), 1.39 (6H, s, CH3 x 2), 1.04 (6H, t, J 7.6, CH2CH3); 1H NMR (MeOD, TFA salt, 400 MHz) δ 10.29 (1H, s, Tetrazine-H), 8.53 (2H, d, J 8.0, Ar-H), 8.09 (2H, d, J 8.0, Ar-H), 7.57 (2H, d, J 8.0, Ar-H), 7.51 (2H, d, J 8.0, Ar-H), 4.50 (2H, m, NHCH x 2), 3.65 (2H, s, CH2), 3.28 (2H, d, J 7.0, CH2), 2.97 (2H, t, J 7.5, CH2), 2.68 (6H, s, CH3 x 2), 2.43 (4H, q, J 7.6, CH2CH3), 2.04 – 1.51 (12H, m, each CH2), 1.37 (6H, s, CH3 x 2), 1.02 (6H, t, J 7.5, CH2CH3); 13C NMR (MeOD, TFA Salt, 100 MHz, CN signals were not observed) δ 175.0, 174.7, 173.2, 169.5 (each C=O), 167.6 (Tetrazine-C), 159.2 (Tetrazine-C), 155.6, 142.6, 141.8, 139.4, 136.8, 136.3, 132.0, 131.2, 130.2, 129.9 (2s), 129.3 (each Ar-C), 55.9, 53.0 (each NHCH), 43.8 (CH2C8H5N4), 40.6, 40.3 (CH2NHCO & CH2NH2), 32.4, 32.0, 30.1, 28.6, 24.4, 23.7 (each CH2), 17.9 (CH2CH3), 14.8 (CH2CH3), 13.5 (CH3), 12.5 (CH3); ESI-HRMS calcd. C48H58BN12O5, 893.4746 found m/z 893.4748 [M-H]-.
2,2',2''-(10-(2-(((S)-5-((S)-6-(2-(4-(1,2,4,5-tetrazin-3-yl)phenyl)acetamido)-2-(4-(4,4-dicyano-2,6-diethyl-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-s-indacen-8-yl)benzamido)hexanamido)-5-carboxypentyl)amino)-2-oxoethyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetic acid (13). To a stirred solution of 12 (14 mg, 14.9 µmol) in dry CH2Cl2 (2 mL) were added NEt3 (22 µL, 157 µmol) and DOTA-NHS (13 mg, 17.3 µmol). The reaction mixture was stirred at room temperature for two hours and the solvent was removed under a stream of air. The resulting residue was purified via a Biotage C-18 column (H2O (0.1% TFA) – ACN (0.1% TFA) 80:20 – 50:50) to give the tile compound 13 (13.5 mg, 52%) at a pink-red solid; Rf 11.2 min (C-18 column, flow rate = 2 mL/min, H2O (0.1% TFA) – ACN (0.1% TFA) 70:30 – 50:50 over 15 min); 1H NMR (CD3CN-D2O, 600 MHz) δ 10.23 (1H, s, Tetrazine-H), 8.40 (2H, d, J 8.4, Ar-H), 7.97 (2H, d, J 8.4 Ar-H), 7.49 (2H, d, J 8.4 Ar-H), 7.39 (2H, d, J 8.5Ar-H), 4.52 (1H, dd, J 9.1, 5.0, NHCH), 4.25 (1H, dd, J 8.0, 5.5 NHCH), 3.70 – 2.95 (30H, m, each CH2), 2.57 (6H, s, CH3 x 2), 2.30 (4H, q, J 7.6, CH2CH3), 1.79 – 1.66 (3H, m, CHaHb & CHa’Hb’), 1.51 – 1.30 (9H, m, CHa’Hb’ & CH2), 1.24 (6H, s, CH3 x 2) 0.90 (6H, t, J 7.6, CH2CH3);1H NMR (MeOD, 600 MHz) δ 10.30 (1H, s, Tetrazine-H), 8.54 (2H, d, J 8.3 Ar-H), 8.09 (2H, d, J 7.8 Ar-H), 7.57 (2H, d, J 8.3 Ar-H), 7.52 (2H, d, J 7.9 Ar-H), 4.56 (1H, dd, J 8.9, 5.5 NHCH), 4.43 (1H, dd, J 9.3, 4.8 NHCH), 4.07 – 2.92 (30H, m, each CH2), 2.68 (6H, s, CH3 x 2), 2.43 (4H, q, J 7.6, CH2CH3), 1.99 – 1.87 (3H, m, CHaHb & CHa’Hb’), 1.79 – 1.73 (1H, m, CHa’Hb’), 1.66 – 1.45 (8H, m, each CH2), 1.37 (6H, s, CH3 x 2), 1.02 (6H, t, J 7.6, CH2CH3); 13C NMR (MeOD, 150 MHz, DOTA signals were not observed) δ 175.4, 174.7, 173.3, 169.4 (each C=O), 167.6 (Tetrazine-C), 161.8 (q, J 36.8, CF3CO2H) 158.7 (Tetrazine-C), 155.5, 142.7, 142.0, 141.5, 139.3, 136.4, 136.1, 132.0, 131.2, 130.1, 129.9, 129.9, 129.3 (each Ar-C), 127.5 (q, J 75, NC-B) 117.6 (q, J 290.2, CF3CO2H), 55.6 (NHCH), 53.5 (NHCH), 44.3 (CH2C8H5N4), 40.3 (2s) (CH2NHCO & CH2NHCO), 32.5, 32.2, 30.1, 29.7, 25.7, 24.3 (each CH2), 17.9 (CH2CH3), 14.9 (CH2CH3), 13.5 (CH3), 12.6 (CH3); ESI-HRMS calcd. C64H84BN16O12, 1279.6542 found m/z 1279.6548 [M+H]+.
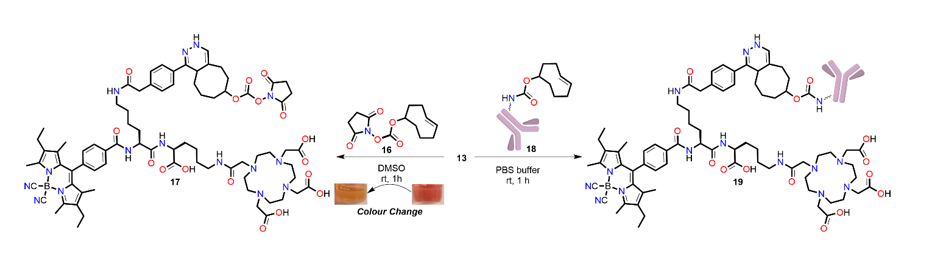
2,2',2''-(10-(2-(((S)-5-carboxy-5-((S)-2-(4-(5-cyano-2,8-diethyl-5-isocyano-1,3,7,9-tetramethyl-5H-4λ4,5λ4-dipyrrolo[1,2-c:2',1'-f][1,3,2]diazaborinin-10-yl)benzamido)-6-(2-(4-((7S,10aR)-7-((((2,5-dioxopyrrolidin-1-yl)oxy)carbonyl)oxy)-3,5,6,7,8,9,10,10a-octahydrocycloocta[d]pyridazin-1-yl)phenyl)acetamido)hexanamido)pentyl)amino)-2-oxoethyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetic acid (17). The stock solution for fluorescent measurements was prepared as follows; 13 (0.3 mg, 0.172 µmol) was dissolved in DMSO (200 µL) then 20 µL of TCO-NHS (1.5 mg, 5.61 µmol in 200 µL DMSO) was added. The reaction mixture was left to stand at room temperature for 1 h. The solution colour changed from pink to orange indicting tetrazine consumption. The product and reaction completion was confirmed via HRMS. ESI-HRMS calcd. C77H101BN15O17, 1518.7593 found m/z 1518.7587 [M+H]+.
Indium complex (14). In a 2 mL centrifuge tube 13 (1.8 mg, 1.04 µmol) was dissolved in MeOH (1 mL). NEt3 (10 µL) and anhydrous Indium chloride (2.3 mg, 10.8 µmol) was added. The reaction mixture was heated at 50 °C for 5 min during which time a precipitate formed. The suspension was centrifuged at 15000 rpm for 2 min, the supernatant was removed and the precipitate was washed with three 2 mL portions of MeOH to give 14 (1.45 mg, quant.) as a red solid; ESI-HRMS calcd. C64H81BInN16O12, 1391.5352 found m/z 1391.5300 [M+H]+.
Lanthanum complex (15). 13 (0.5 mg, 0.28 µmol) was dissolved in sodium acetate buffer (1 mL, 100 mM pH = 6) and Lanthanum chloride hydrate (1 mg, 4.08 µmol) was added. The reaction mixture was heated at 45 °C for 60 min during which time a precipitate formed. The suspension was transferred to a 2 mL centrifuge tube and centrifuged at 15000 rpm for 2 min. The supernatant was removed and the precipitate was washed with three 2 mL portions of water. Lyophilisation gave 15 (0.4 mg, quant.) as a red solid; ESI-HRMS calcd. C64H81BLaN16O12, 1415.5376 found m/z 1415.5389 [M+H]+.
Preparation of trastuzumab-TCO and conjugation with DO3A-BODIPY-Tz
TCO conjugation to trastuzumab. Procedures followed closely those previously published.23 Purified trastuzumab (Herceptin) (5.0 mg, 47.9 mg/mL) in PBS buffer (pH 7.4) was adjusted to pH 8.5 using small aliquots (1-5 µL) of Na2CO3 solution (0.1 M) in a 1.5 mL Eppendorf tube. To the antibody solution, TCO-NHS (40 mg/mL solution in DMF, 40 mol equiv.) was added slowly with agitation. The reaction mixture was incubated at 25 °C on a thermomixer for 1 h with mild agitation (500 rpm), and subsequently purified using PD-10 desalting columns (GE Healthcare) and collected in PBS (pH 7.4, 2 x 1 mL). The concentration of trastuzumab-TCO was measured using a Nanodrop UV-Vis spectrophotometer monitoring the 280 nm wavelength (ɛ280 = 210,000 M-1cm-1), and the number of TCO moieties per antibody was determined by MALDI-ToF MS/MS to be 4.3.
DOTA-BODIPY-Tz and trastuzumab-TCO in vitro “click”. To a solution of trastuzumab-TCO (100 μg in 500 μL PBS), DOTA-BODIPY-Tz (13) (35 μL of 1.3x10-3 M solution in DMSO, ~70 equiv.) was added slowly with agitation. The reaction mixture was rotated at ambient temperature for 1 hour, and subsequently purified using PD-10 desalting columns (GE Healthcare) and collected in PBS (pH 7.4, 2 mL). The number of DOTA-BODIPY-Tz moieties ‘clicked’ per antibody was determined by MALDI-ToF MS/MS to be 2.9.
111In & 225Ac Radiolabeling and Radiometal Complex Stability Studies
General Methods and Instrumentation. [111In]InCl3 was purchased from BWXT (Vancouver, BC, Canada) and received as ~0.05 N HCl solution. [225Ac]Ac(NO3)3 was produced via the spallation of thorium targets on TRIUMF’s 500 MeV cyclotron (Vancouver, BC, Canada), and isolated as previously described51 in dilute acid (~0.05 M HNO3). Aluminum-backed TLC plates (silica gel 60, F254, EMD Millipore) or paper-backed instant TLC plates (silica gel, iTLC-SG, Agilent) were used to analyze 225Ac or 111In radiolabeling reaction progress, respectively. TLC plates were developed and then were measured on a BioScan System 200 imaging scanner equipped with a BioScan Autochanger 1000 and WinScan software and radiolabeling yields were calculated by integrating the peaks in the radio-chromatogram. For 225Ac radiolabeling, developed plates were counted at least 8 hours later to allow for daughter isotopes to decay completely, to ensure the radioactive signal was generated solely by parent 225Ac.
The radioactive RP-HPLC system used to analyse 111In radiolabeling yields consisted of a Phenomenex Luna C18(2) 100 Å RP analytical column (5 μm, 100 x 4.6 mm) using an Agilent HPLC equipped with a model 1200 quaternary pump, a model 1200 UV absorbance detector (set at 250 nm) and a Raytest Gabi Star NaI(Tl) radiation detector.
111In radiolabeling studies. Radiolabeling procedures followed closely those outline previously.52–54 Briefly, DO3A-BODIPY-Tz (13·4TFA) was made up as a stock solution (1 mg mL-1, 5.76 x 10-3 M) in DMSO. Using serial dilution, stock ligand solutions with concentrations of 5.76 x 10-4 – 10-6 M were also prepared in deionized water. A 10 µL aliquot of each ligand stock solution (or 10 µL of deionized water as a blank) was added to a Eppendorf tube and diluted with ammonium acetate buffer (0.15 M, pH 5) such that the final reaction volume was 100 µL after the addition of [111In]InCl3, to give final ligand concentrations of 5.76 x 10-4 – 10-7 M. An aliquot of [111In]InCl3 (3.7 – 37 MBq) was added to the Eppendorf tubes containing ligand and buffer and allowed to react for 30 – 60 min at 45°C. Reaction progress was analyzed at 30 and 60 min by spotting a small aliquot (1 – 3 µL) onto the bottom of an instant thin layer chromatography (iTLC-SG) plate and developed using mobile phase (MP) A (EDTA, 50 mM, pH 5). Under these conditions, uncomplexed [111In]In3+ travels with the solvent front (Rf ~ 1) while [111In]In-complexed species stick to the baseline (Rf = 0). Alternatively, radiolabeling yields were determined by analytical RP-HPLC. Elution conditions used for RP-HPLC analysis were gradient: A: 0.1 % TFA in water; B: 0.1% TFA in acetonitrile; 0 to 100% B linear gradient 20 min, 1 mL min-1. [111In][In-DO3A-BODIPY-Tz] (tR = 11.6 min), “free” 111In3+ (tR = 1.56 min). According to the radiolabeling yield observed, the 111In-complexes were either purified using a C18-light cartridge (Waters Sep-pak, pre-conditioned with 5 mL ethanol, then 10 mL of labeling buffer) eluted with 300 μL of ethanol or used without further purification.
Conjugation of [111In]In-DO3A-BOPIDY-Tz to trastuzumab-TCO. The [111In]In-DO3A-BOPIDY-Tz chelation reaction was mixed with a solution of trastuzumab-TCO (200 µg, in 200 µL PBS pH 7.4). The reaction mixture was agitated at ambient temperature, the progress of the radioconjugation was determined by iTLC-SG developed using MP B: ethanol/water (50:50). Under these conditions, [111In]In-DO3A-BOPIDY-Tz travels up the plate (Rf ~ 0.5) while the conjugated antibody [111In]In-DO3A-BOPIDY-trastuzumab remains at the baseline (Rf = 0). The reaction mixture was purified by passage over a PD-10 desalting column (GE Healthcare) using PBS (pH 7.4) as mobile phase. Radiochemical purity was analyzed by iTLC-SG using MP B.
225Ac radiolabeling studies. Radiolabeling procedures followed closely those outline previously.19,40 A 1x10-3 M stock solution of DO3A-BODIPY-Tz (13·4TFA) was prepared in DMSO, and serial dilutions in deionized water were prepared to give additional solutions of 10-4 and 10-5 M. A 10 µL aliquot of each ligand stock solution (or 10 µL of deionized water as a blank) was added to a Eppendorf tube in triplicate and diluted with ammonium acetate buffer (0.15 M, pH 7) such that the final reaction volume was 100 µL after the addition of [225Ac]Ac(NO3)3, to give final ligand concentrations of 1 x 10-4 – 10-6 M. An aliquot of [225Ac]Ac(NO3)3 (37-100 kBq) was added to the Eppendorf tubes containing ligand and buffer and allowed to react for 30 – 60 min at 80°C. Reaction progress was analyzed at 30 and 60 min by spotting a small aliquot (1 – 3 µL) onto the bottom of an aluminum-backed TLC silica gel plate and developed using MP C (citric acid, 0.4 M, pH 4). Under these conditions, uncomplexed [225Ac]Ac3+ travels with the solvent front (Rf ~ 1) while [225Ac]Ac-complexed species stick to the baseline (Rf = 0). According to the radiolabeling yield observed, the 225Ac-complexes were either purified using a C18-light cartridge (Waters Sep-pak; pre-conditioned with 5 mL ethanol, then 10 mL of labeling buffer) eluted with 300 μL of ethanol or used without further purification.
Conjugation of [225Ac]Ac-DO3A-BOPIDY-Tz to trastuzumab-TCO. The [225Ac]Ac-DO3A-BOPIDY-Tz chelation reaction (containing either 17 or 1.7 μg of ligand), taken directly after radiolabeling or post-C18 purificaion, was mixed with varying amounts of trastuzumab-TCO (748 – 14.7 µg, in PBS pH 7.4) to give ligand-to-antibody ratios of 2:1, 4:1, and 10:1; the reaction volume was set to 1 mL by the addition of PBS (pH 7.4). The reaction mixture was agitated at 37°C, the progress of the radioconjugation was determined by iTLC-SG developed using MP B: ethanol/water (50:50). Under these conditions, [225Ac]Ac-DO3A-BOPIDY-Tz travels up the plate (Rf ~ 1) while the conjugated antibody [225Ac]Ac-DO3A-BOPIDY-trastuzumab remains at the baseline (Rf = 0). The reaction mixture was purified by passage over a PD-10 desalting column (GE Healthcare) using PBS (pH 7.4) as mobile phase. Radiochemical purity (%RCP) was analyzed by iTLC-SG using MP B.
Radiometal-complex stability studies in human serum. Pre-formed [111In]In- and [225Ac]Ac-DO3A-BODIPY-Tz species (RCP >99%) or radiolabeling controls (water was substituted for ligand) were incubated in human serum (1:1 volume based on labeling reaction volume), and the solutions were agitated (450 rpm) at 37 °C. The solutions were monitored over the course of 5 – 6 days by TLC. For competition studies with 111In, iTLC-SG plates using MP B was employed. Under these conditions, [111In]In-DO3A-BODIPY-Tz travels up the plate (Rf ~ 1), while 111In3+ that has transchelated to serum proteins remains at the baseline (Rf = 0). For competition studies with 225Ac, aluminum-backed TLC silica gel plates using MP C was employed. Under these conditions, [225Ac]Ac-DO3A-BODIPY-Tz remains at the baseline (Rf ~ 0), while uncomplexed 225Ac3+ that has detached from the ligand travels with the solvent front (Rf = 1).
Preparation of [111In]In-DO3A-BODIPY-Trastuzumab for in vivo studies. To a solution of DO3A-BODIPY-Tz (9.25 μg) in ammonium acetate (0.1 M, pH 5.5), was added [111In]InCl3 (272 MBq in 0.01 M HCl), such that the final reaction volume was 100 μL. The reaction mixture was reacted with agitation (400 rpm) at 45 °C for 45 min. Radiochemical conversion yield (%RCC) was determined to be > 99% by iTLC-SG (0.5 μL spot) using MP A. The radiolabeling reaction was added directly to a solution of trastuzumab-TCO (400 μg, 2:1 ligand-to-mAb ratio) in PBS (900 μL, pH 7.4), and reacted with agitation (400 rpm) at 37°C for 60 min. The radioconjugation progress was assessed by iTLC-SG using MP B, and the reaction mixture was subsequently purified over a PD-10 desalting column (GE Healthcare) and collected in 2.0 mL of PBS (pH 7.4). The radiochemical purity (RCP) was determined to be >99% by iTLC-SG using MP B. The specific activity of the final radiotracer (0.3 MBq/μg) was determined by measuring the activity in a dose calibrator and considering a recovery of 80% of the mAb after the PD-10 desalting column.
In vivo and Ex vivo Biodistribution and immuno-SPECT Imaging Studies
SKOV-3 Xenograft Mouse Model. All experiments were conducted in accordance with the guidelines established by the Canadian Council on Animal Care and approved by the Animal Ethics Committee of the University of British Columbia (protocol no. A20-0113). Female nude mice (8 weeks old) obtained from Jackson laboratory (stock#002019 from JAX) were subcutaneously injected with 8 x 106 SKOV-3 cells in Matrigel (BD Bioscience, 1:1 PBS:matrigel) on the left shoulder.
In vivo biodistribution and immuno-SPECT imaging. Mice bearing SKOV-3 ovarian cancer xenografts were administered with 4.1 ± 0.1 MBq (26.3 ± 0.3 µg; n = 4) of [111In]In-DO3A-BODIPY-Tz-TCO-trastuzumab in ~100 µL of PBS (pH 7.4) via tail-vein injection. Mice were imaged 1, 3, or 5 days after injection. Image acquisition and reconstruction was performed using the U-SPECT-II-CT (MILabs, Utrecht, The Netherlands). Prior to image acquisition, mice were anesthetized via inhalation of 2% isoflurane-oxygen gas mixture and placed on the scanner bed with a heating pad to maintain body temperature. A 5 min CT scan was obtained for localization with voltage setting at 60 kV and current at 615 µA followed by a static emission scan using an ultrahigh-resolution multipinhole rate-mouse (1 mm pinhole size) collimator. Data were acquired in list mode, reconstructed using the U-SPECT II software, and co-registered for alignment. SPECT images were reconstructed using maximum likelihood expectation maximization (3 iterations), pixel-based ordered subset expectation (16 subsets), and a postprocessing filter (Gaussian blurring) of 0.5 mm centered at photopeaks 171 and 245 keV with a 20% window width. Imaging data sets were decay corrected to injection time, and converted to DICOM data for visualization in the Inveon Research Workplace (Siemens Medical Solutions USA, Inc.). For biodistribution studies, at 6 days post injection, mice were sacrificed by the inhalation of isoflurane followed by CO2, blood was withdrawn by cardiac puncture, and tissues of interest including fat, uterus, ovaries, intestine, spleen, liver, pancreas, stomach, adrenal glands, kidney, lungs, heart, SKOV-3 tumor, muscle, bone, and brain were harvested, washed in PBS, dried, and weighed. Activity of each sample was measured by a calibrated γ counter (PerkinElmer, Wizard 2 2480) with decay correction. The activity uptake was expressed as percentage of injected dose per gram of tissue (% ID/g).
Ex vivo autoradiography. Half of the SKOV-3 tumors were harvested and frozen in a cryoprotective gel (Tissue-Tek optimal cutting temperature compound, Sakura) using dry ice for autoradiography and histology. They were then cut using the NX70 cryostat (Thermo Fisher Scientific) set at a temperature of -15ºC, mounted on Superfrost Plus Gold slides and fixed in methanol for 5min at room temperature. For autoradiography, a phosphor screen was applied on 14μm-thick sections and the resulting signals were acquired with the PhosphoImager (GE Typhoon FLA 9500). The consecutive sections of 8 µm were stained with Hematoxylin and Eosin (H&E) following supplier recommendations (Leica Biosystem).
{kind=link}
{kind=link}