All reagents and chemicals were purchased from sigma Aldrich, Fluka, BDH and used as received. The 1H and 13C NMR spectra were recorded in DMSO-d6 or CDCl3-d on Bruker biospin ICON-NMR spectrometer and AVANCE AV-300 spectrometer (US) using TMS as internal reference. The apparent resonance multiplicity is described as s (singlet), bs (broad singlet), d (doublet), dd (doublet of doublet), t (triplet), q (quartet) and m (multiplet). Infrared measurements were recorded in 400-4000cm− 1 on a spectrum 2000 FTIR spectrophotometer by Perkin Elmer (USA). Melting point was determined in a capillary tube using a Gallenkamp (UK) electrothermal melting point apparatus. The enantiomeric excess was determined by chiral column, Lux 5µm Cellulose-1, LC Column 250x4.6 mm (USA). The instrument used for this technique was HPLC PerkinElmer (USA). Analysis of sample with an evaporation point of max. 300oC; mass range: m/z 30–800 amu. VG Instruments autospec/EBEE-Geometry was used to record mass spectra. Electron impact (EIMS) yield mostly fragment ions. Molecular ions are not always observed. High resolution features with ca 6000–8000 resolution. CD spectra were measured by JASCO-815 CD spectrometer (USA) in static mode. For CD measurements, the warfarin analogues were dissolved in a mixture of aqueous phosphoric acid and acetonitrile having ratio 4:6 and pH 2.
General procedure for synthesis of analogues of 4-hydroxy-3-(3-oxo-1-phenylbutyl)-2H-chromen-2-one
Scheme 1 presented complete synthetic strategies. In first step α,β unsaturated ketones (3a-3k) (chalcones) were synthesized[32,33] by taking 1:1 mole ratio of substituted aldehyde (1a-1e) and substituted ketone (2a-2f) in a two neck round bottom flask equipped with magnetic stirrer and dissolve it in minimum amount of ethanol/methanol at room temperature and then add 10% NaOH drop wise in above solution and leave for 2hrs at room temperature and at the end of reaction neutralized by dilute HCl and extracted by ethanol and dried in a rotary evaporator. Solid product obtained was purified by column chromatography by using 7:3 ratio of n-Hexane and ethyl acetate respectively and recrystallization in ethanol and their structures were confirmed by the comparison of their melting points and IR data with the reported values.
Then to prepare warfarin analogues 5a-5k; 4-hydroxycoumarin (0.32mmol), α,β unsaturated ketones (0.2mmol), and 20mol % 9-amino-9-deoxy epiquinine were dissolved in 20ml of dry DCM in a two neck round bottom flask equipped with magnetic stirrer, followed by addition of 40 mol % trifluoracetic acid (TFA) as additive.[24] The reaction mixture was stirred at room temperature for 3 to 4 days at room temperature and the progress of reactions was monitored by TLC visualized under UV lamp and developed in vanillin spray. The crude products were purified by column chromatography using different ratios of n-hexane and ethyl acetate. A single spot of product was obtained on TLC after column chromatography and purity was further verified by HPLC.
Chiral HPLC
Enantiomeric excess was determined by chiral stationary phase HPLC using Lux 5 µm cellulose − 1, LC column 250x4.6 mm. Mixture of n-hexane and isopropanol in ratio of 60:40 with 0.1% formic acid was used as eluent using flow rate of 1ml/min and data acquisition time was 10min.
Enantiomeric excess (% ee) =\(\frac{\left[S\right]-\left[R\right]}{\left[S\right]+\left[R\right]}X100\)
Circular Dichroism Studies
CD spectra were measured by JASCO-815 CD spectrophotometer in static mode. For CD measurements, 200µg/ml of samples were prepared by dissolving synthesized compounds in a mixture of aqueous phosphoric acid and acetonitrile (4:6) having pH 2.0.
As regards the measuring parameters, wavelength range was 170-400nm, data interval time 1sec, response time 2sec, spectral band width 1nm, number of accumulations 3, and optical path length 10mm were used as provided in JASCO CD spectrometer’s instrumental manual for CD measurement of warfarin.
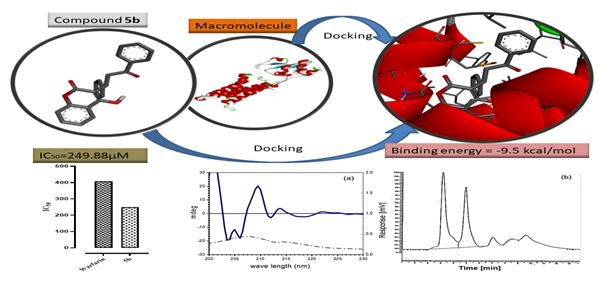
All the synthesized products showed maximum UV absorbance band between 190 to 220nm. In CD, the cotton effect of the band at the value of the maximum absorption of UV was compared with the reported cotton effect of warfarin provided in JASCO CD spectrometer’s instrumental manual. A negative cotton effect in CD in particular absorption indicates the (S) enantiomer in excess. The CD spectra are given in Fig. 8.
Tautomerism
The percentage of keto-hemiketal tautomers was determined by following the already reported method.[34] Briefly, the 1HNMR peaks integration from simple one dimensional spectra are used to determine the ratio of tautomers. The percentages of the keto-hemiketal tautomers is presented in Table 3.
Table 3
The keto-hemiketal percentage of synthesized warfarin analogues
|
Compound
|
Solvent
|
Keto%
|
Hemiketal%
|
|
5a
|
CDCl3
|
64
|
36
|
|
5b
|
CDCl3
|
69
|
31
|
|
5c
|
CDCl3
|
52
|
48
|
|
5d
|
CDCl3
|
85
|
15
|
|
5e
|
CDCl3
|
60
|
40
|
|
5f
|
DMSO
|
62
|
38
|
|
5g
|
CDCl3
|
43
|
53
|
|
5h
|
CDCl3
|
49
|
51
|
|
5i
|
CDCl3
|
60
|
40
|
|
5j
|
CDCl3
|
54
|
46
|
|
5k
|
CDCl3
|
51
|
49
|
4-hydroxy-3-[1-(4-hydroxy-3-methoxyphenyl)-3-oxobutyl]-2H-chromen-2-one (5a)
Solid; Yield 90%; mp 139-141oC; ee 91% [S]; IR (acetone) vmax 1720 (C = O), 1625 (C = O), 1510 (C-H), 1380 (O-H) cm− 1; 1H NMR (CDCl3, 200 MHz) δ 8.69 (1H, bs, OH), 7.80 (1H, m, ArH), 7.43 (1H, m, ArH), 7.20 (2H, m, ArH), 6.88 (3H, m, ArH), 5.46 (1H, s, OH), 4.08 (2H, m, CH2 Keto), 3.77 (3H, s, OCH3), 2.35 (1H, m, CH of steric center), 1.66 (2H, s, CH2); 13C NMR (CDCl3, 50 MHz) δ 162.1 (C = O), 161.2 (C = O), 159.6 (C-OH), 158.6 (ArC), 152.9 (ArC), 145.0 (ArC), 135.0 (ArC), 132.9 (ArC), 132.0 (ArC), 123.9 (ArC-OH), 123.6 (ArC), 122.74 (ArC), 117.0 (ArC), 116.6 (ArC), 115.0 (ArC), 114.0 (ArC), 111.1 (ArC), 99.0 (CH), 60.4 (CH2 Keto), 56.0 (CH), 42.7 (CH2), 39.8 (CH3), 33.6, 29.6 (CH); EI MS m/z [M+ 1] 354.1 [C20H18O6+] (40%).
4-hydroxy-3-(3-oxo-1,3-diphenylpropyl)-2H-chromen-2-one (5b)
Solid; Yield 80%; mp150oC; ee 16% [S]; IR (acetone) vmax 1741 (C = O), 1677 (C = O), 1570 (C-H), 1380 (O-H) cm-1; 1H NMR (CDCl3, 200 MHz) δ 9.78 (1H, s, OH), 8.03 (2H, m, ArH), 8.00 (1H, s, ArH), 7.94 (1H, d, J = 1.7 Hz, ArH), 7.90 (1H, d, J = 1.7 Hz, ArH), 7.54 (1H, m, ArH), 7.46 (1H, d, J = 1.4 Hz, ArH), 7.42 (1H, q, J = 1.7 Hz, ArH), 7.38 (1H, m, ArH), 7.32 (1H, s, ArH), 7.27 (1H, m, ArH), 7.24 (2H, d, J = 1.0 Hz, ArH), 7.14 (1H, m, Ar H), 4.86 (1H, d, J = 2.4 Hz, CH2), 4.05 (1H, m, CH of steric center), 3.77 (1H, d, J = 2.4 Hz, CH2), 3.68 (1H, d, J = 2.4 Hz, CH); 13C NMR (CDCl3, 50 MHz) δ 161.1 (C = O), 152.9 (C = O), 139.9 (C-OH), 134.42 (C-OH), 131.7 (ArC), 128.9 (ArC), 128.7 (ArC), 128.2 (ArC), 126.7 (ArC), 124.0 (ArC), 116.2 (ArC), 77.2 (CH), 45.1 (CH), 35.2 (CH), 29.6 (CH2); EI MS m/z [M+ 1] 370.2 [C24H18O4+] (15%).
4-hydroxy-3-(1-(4-hydroxy-3-methoxyphenyl)-3-oxo-3-phenylpropyl)-2H-chromen-2-one (5c)
Solid; Yield 68%; m.p. 154oC; ee 70% [S]; IR (acetone) vmax 1743 (C = O), 1607 (C = O), 1513 (C-H), 1360 (O-H) cm-1; 1H NMR (CDCl3, 200 MHz) δ 9.76 (1H, s, OH), 7.66 (1H, s, ArH), 7.63 (1H, s, ArH), 7.64 (1H, s, ArH),7.62 (1H, s, ArH), 7.61 (2H, s, ArH), 7.48 (2H, s, ArH), 7.46 (2H, s, ArH), 7.45 (1H, s, ArH), 7.35 (1H, s, ArH), 5.35 (1H, s, Ar-OH), 4.47 (1H, d, CH2, J = 4.0 Hz), 3.83 (1H, d, CH, J = 6.0 Hz ), 3.80 (3H, s, OCH3), 3.76 (1H, m, CH), 3.55 (1H, d, J = 6.0 Hz, CH); 13C NMR (DMSO, 50 MHz) δ 206.5 (C = O), 165.7 (C = O), 161.9 (C-OH), 153.5 (ArC), 132.7 (ArC), 123.9 (ArC), 123.2 (ArC), 116.3 (ArC), 115.8 (ArC), 90.8 (CH), 56.0 (CH), 55.5 (CH2), 30.6 (OCH3), 19.6 (CH); EI MS m/z [M+ 1] 416.1 [C25H20O6+] (5%).
4-hydroxy-3-(3-(naphthalen-3-yl)-3-oxo-1-phenylpropyl)-2H-chromen-2-one (5d)
Solid; Yield 60%; mp 210 ºC; ee 60% [S]; IR (acetone) vmax 1715 (C = O), 1670 (C = O), 1508 (C-H), 1384 (O-H) cm-1; 1H NMR (CDCl3, 200 MHz) δ 9.88 (1H, s, OH), 8.18 (1H, d, ArH, J = 1.8 Hz), 8.05 (1H, m, ArH), 7.86 (1H, d, ArH J = 6.0 Hz), 7.79 (2H, d, J = 5.3 Hz, ArH), 7.69 (1H, dd, J1 = 8.7 Hz, J21.8 Hz, ArH), 7.55 (1H, m, ArH), 7.37 (2H, q, ArH, J = 1.6 Hz), 7.31 (1H, m, ArH), 7.27 (2H, q, J = 1.5 Hz, ArH ), 7.23 (1H, m, ArH), 5.92 (1H, d, J = 5.0 Hz, CH2), 4.70 (1H, d, J = 4.9 Hz, CH2), 2.10 (1H, s, CH), 1.17 (1H, m, CH, steric center); 13C NMR (CDCl3, 50 MHz) δ 167.1 (C = O), 133.4 (C-OH), 132.9 (ArC), 129.7 (ArC), 128.6 (ArC), 127.1 (ArC), 126.6 (ArC), 123.9 (ArC), 122.6 (ArC), 77.1 (CH), 36.7 (CH2), 32.4 (CH2). 27.28 (CH); EIMS peak m/z 403.1 [C27H15O4+] (20%), 402.1 [C27H14O4+] (65%), 403.1 [C27H15O4+] (20%), 326.1 [C21H10O4+] (28%), 325.1 [C21H9O4+] (100%), 282.1 [C17H14O4+] (16%), 265 [C17H13O3+] (3%), 252.1 [C16H12O3+] (12%), 176.1 [C11H12O2+] (6%), 127 [C6H7O3+] (14%), 121 [C7H5O2+] (12%), 93 [C6H5O+] (3%), 77.1 [C6H5+] (6%), 51.1 [C4H3+] (4%), 26.9 [C2H3+] (6%).
4-hydroxy-3-(1-(4-hydroxy-3-methoxyphenyl)-3-(2-hydroxyphenyl)-3-oxopropyl)-2H-chromen-2-one (5e)
Solid; Yield 75%; mp 192ºC; ee 51% [S]; IR (acetone) vmax 1700 (C = O), 1620 (C = O), 1480 (C-H), 1390 (O-H) cm-1; 1H NMR (CDCl3, 200 MHz) δ 7.94 (3H, d, J = 8.4 Hz, ArH), 7.64 (1H, m, ArH), 7.54 (1H, dd, J = 7.1 1.5 Hz, ArH), 7.47 (1H, m, ArH), 7.35 (1H, d, J = 5.3 Hz, ArH), 7.32 (2H, s, ArH), 6.80 (1H, d, J = 8.2 Hz, ArH), 6.65 (1H, m, ArH), 6.0 (1H, s, OH), 5.25 (1H, m, CH), 4.26 (2H, d, J = 4.3 Hz, CH2), 4.21 (1H, s, OH), 4.15 (1H, dd, J1 = 1.7 Hz, J2 = 5.9 Hz, CH), 4.09 (1H, s, Ar-OH), 3.79(1H, d, CH), 3.68 (3H, s, OCH3); 13C NMR (CDCl3, 50 MHz): δC 200.4 (C = O), 166.0 (C = O), 162.2 (C-OH), 161.7 (C-OH), 148.3 (ArC), 146.7 (ArC), 146.6 (ArC), 135.8 (ArC), 135.3 (ArC), 132.5 (ArC), 130.2 (ArC),124.5 (ArC), 124.4 (ArC), 121.6 (ArC), 120.8 (ArC), 120.5 (ArC),116.3 (ArC), 116.1 (ArC), 112.2 (ArC), 102.5 (CH), 56.2 (OCH3), 47.1 (CH), 38.1(CH2); EI MS m/z [M+ 1] 432.1 [C25H20O7+] (3%).
3-[3-(4-aminophenyl)-3-oxo-1-phenylpropyl]-4-hydroxy-2H-chromen-2-one (5f)
Solid; Yield 80%; mp 137 ºC; ee 29% [S]; IR (acetone) vmax 3400 (N-H), 1700 (C = O), 1610 (C = O),1585 (N-H), 1495 (C-H), 1300 (O-H) cm− 1; 1H NMR (DMSO, 200 MHz) δ 7.86 (2H, d, J = 1.7 Hz, ArH), 7.82 (2H, d, J = 1.9 Hz, ArH), 7.66 (2H, ddd, J1 = 8.6 Hz, J2 = 7.1 Hz, J3 = 1.7 Hz, ArH), 7.39 (4H, m, ArH), 7.27 (2H, m, ArH), 7.11 (1H, m, ArH), 5.61 (2H, s, NH2), 2.24 (1H, m, CH), 2.22 (1H, m, CH, steric center), 2.0 (1H, m, OH), 1.25 (1H, d, CH2, J = 7.2 Hz), 1.09 (1H, m, CH2); 13C NMR (DMSO, 50 MHz,) δ 167.5 (C = O), 165.6 (C = O), 161.9 (C-OH), 153.5 (ArC-NH2), 152.4 (C-OH) 132.7 (ArC-OCH3), 123.9 (ArC), 123.2 (ArC),119.8 (ArC), 116.3 (ArC), 115.7 (ArC-OH), 103.96 (ArC), 90.9 (C-H), 20.7 (CH2), 14.4 (CH); EI MS peak m/z 367.1 [C24H17O3N+] (9%), 290.1 [C19H16O2N+] (16%), 223.1 [C14H511O2N+] (14%),162 [C9H6O3+] (43%) 121 [C7H5O2+] (12%), 120.0 [C7H4O2+] (100%), 93 [C6H7N+] (12%), 92 [C6H6N+] (71%), 77.1 [C6H5+] (15%), 51.0 [C4H3+] (14%), 26.9 [C2H3+] (6%).
3-[3-(4-chlorophenyl)-3-oxo-1-phenylpropyl]-4-hydroxy-2H-chromen-2-one (5g)
Solid; Yield 76%; mp166 ºC; ee 54% [S]; IR (acetone) vmax 1720 (C = O), 1625 (C = O), 1500 (C-H), 1300 (O-H) cm− 1; 1H NMR (CDCl3, 200 MHz,) δ 9.52 (1H, s, OH), 7.97 (1H, t, J = 1.9 Hz, ArH), 7.91 (2H, m, ArH), 7.66 (1H, t, ArH, J = 3.4 Hz), 7.62 (1H, d, ArH, J = 3.4 Hz), 7.49 (1H, d, ArH, J = 4.0 Hz), 7.45 (1H, d, ArH, J = 2.4 Hz), 7.42 (2H, t, J = 1.9 Hz, ArH), 7.38 (1H, d, J = 1.6 Hz, ArH), 7.26 (2H, m, ArH), 7.14 (1H, m, ArH), 4.86 (1H, dd, J1 = 9.9 Hz, J2 = 2.5 Hz, CH2), 4.37 (1H, m, CH, steric center ), 3.68 (1H, dd, J1 = 19.1 Hz, J2 = 2.6 Hz, CH2), 2.28 (1H, s, OH), 1.60 (1H, d, J = 6 Hz, CH); 13C NMR (CDCl3, 50 MHz): δC 201.2 (C = O), 167.7 (C = O), 152.8 (C-OH), 135.2 (ArC), 132.5 (ArC), 132.2 (ArC), 131.8 (ArC), 130.8 (ArC), 130.0 (ArC), 129.2 (ArC), 128.8 (ArC), 128.2 (ArC), 128.0 (ArC), 116.2 (C-H), 68.2 (CH2), 38.8 (CH), 22.9 (CH); EI MS m/z [M+ 1] 404.2 [C24H17O4Cl+] (5%).
3-(3-(4-chlorophenyl)-1-(3,4,5-trimethoxyphenyl)-3-oxopropyl)-4-hydroxy-2H-chromen-2-one (5h)
Solid; Yield 65%; mp 110 ºC; ee 41% [S]; IR (acetone) vmax 1725 (C = O), 1600 (C = O), 1510 (C-H), 1330 (O-H) cm-1; 1H NMR (CDCl3, 200 MHz) δ 7.95 (1H, m, ArH), 7.65 (1H, t, J = 3.0 Hz, ArH), 7.61 (1H, d, J = 2.5 Hz, ArH), 7.46 (1H, m, ArH), 7.37 (1H, dd, J1 = 8.6 Hz, J2 = 3.8 Hz, ArH), 7.29 (1H, s, ArH), 7.13 (1H, m, ArH), 7.03 (1H, m, ArH), 6.29 (1H, s, ArH), 6.12 (1H, s, ArH), 4.16 (1H, d, J = 1.6 Hz, CH2), 4.14 (1H, d, J = 1.8 Hz, CH2), 3.85 (3H, s, OCH3), 3.69 (1H, m, OH), 3.63 (1H, m, OCH3), 3.58 (1H, s, CH, steric center), 3.45 (1H, d, J = 2.6 Hz, CH), 3.44 (3H, s, OCH3); 13C NMR (CDCl3, 50 MHz) δ 206.9 (C = O), 167.7 (C = O), 153.0 (C-OH), 152.9 (ArC), 134.3 (ArC-Cl), 132.5 (ArC), 131.5 (ArC), 130.9 (ArC), 129.4 (ArC), 128.8 (ArC), 128.7 (ArC), 128.50 (ArC), 126.55 (ArC), 107.4 (ArC), 106.2 (CH), 105.2 (ArC), 68.2 (CH2), 56.2 (CH), 56.16 (OCH3), 55.82 (OCH3), 38.7 (OCH3), 30.4 (OCH3), 22.9 (OCH3); EIMS peak m/z 383.5 [C21H16O5Cl+] (2%), 279.2 [C18H15O3+] (6%), 212.1 [C14H12O2+] (18%), 167.1 [C12H7O+] (28%), 149.0 [C9H9O2+] (100%), 121.1 [C7H5O+] (10%), 71[C4H7O+] (21%), 57 [C3H5O+] (35%), 43[C2H3O+] (24%).
3-(1,3-bis(4-chlorophenyl)-3-oxopropyl)-4-hydroxy-2H-chromen-2-one (4i)
Solid; mp 122 ºC; Yield 62%; ee 24% [S]; IR (acetone) vmax 1710 (C = O), 1625 (C = O), 1500 (C-H), 1360 (O-H) cm-1; 1H NMR (CDCl3, 200 MHz) δ 9.64 (1H, s, OH), 7.96 (1H, d, J = 1.9 Hz, ArH), 7.93 (2H, d, J = 2.0 Hz, ArH), 7.89 (1H, d, J = 1.7 Hz, ArH), 7.42 (2H, d, J = 2.1 Hz, ArH), 7.29 (1H, d, J = 2.2 Hz, ArH), 7.26 (2H, d, J = 2.6 Hz, ArH), 7.22 (1H, s, ArH), 7.12 (1H, m, ArH), 5.19 (1H, dd, J1 = 10.2 Hz, J2 = 1.6 Hz, CH2), 4.80 (1H, dd, J1 = 10.2 Hz, J2 = 2.3 Hz, CH2), 4.27 (1H, m, CH, steric center), 3.67 (1H, d, J = 2.3 Hz, CH); 13C NMR (CDCl3, 50 MHz) δ 201.1 (C = O), 173.28 (C = O), 165.2 (C-OH), 157.8 (ArC), 147.3 (ArC-Cl), 133.1 (ArC-Cl), 131.9 (ArC), 130.0 (ArC), 129.5 (ArC), 129.2 (ArC), 128.2 (ArC), 123.9 (ArC), 117.5 (ArC), 116.2 (ArC), 62.11(CH2), 34.0 (CH), 29.5 (CH), 14.10 (CH); EIMS peak m/z 429.1 [C23H18O4Cl2+] (3%), 383.4 [C22H20O4Cl+] (3%), 265 [C17H13O3+] (2%), 183 [C13H11O+] (10%), 167 [C9H11O3+] (30%), 149.0 [C9H9O2+] (100%),121 [C11H7O3+] (8%), 93 [C6H5O+] (12%), 71 [C4H7O+] (33%), 57 [C3H5O+] (60%).
3-(3-(4-chlorophenyl)-1-(4-methoxyphenyl)-3-oxopropyl)-4-hydroxy-2H-chromen-2-one (5j)
Solid; As product was sticky so melting point cannot be determined. Yield 60%; ee 99.98% [S]; IR (acetone) vmax 1745, 1600, 1480, 1300 cm-1; 1H NMR (CDCl3, 200 MHz) δ 7.71 (1H, s, ArH), 7.65 (2H, d, J = 3.3 Hz, ArH), 7.64 (1H, s, ArH), 7.62 (2H, d, J = 3.3 Hz, ArH), 7.55 (1H, d, J = 19.5 Hz, ArH), 7.48 (2H, s, ArH), 7.46 (2H, s, ArH), 7.45 (1H, s, ArH), 7.43 (1H, s, ArH), 5.26 (1H, d, J = 5.3 Hz, CH2), 5.22 (1H, d, J = 1.5 Hz, CH2), 5.19 (1H, d, J = 1.6 Hz, CH), 5.16 (1H, m, CH, steric center), 4.16 (3H, s, OCH3), 4.03 (1H, bs, OH). 13C NMR (CDCl3, 50 MHz): δC 203.5 (C = O), 173.3 (C = O),167.73 (C-OH), 162.3 (C = O), 137.0 (ArC), 132.4 (ArC), 130.8 (ArC-Cl), 128.8 (ArC-OCH3), 121.8 (ArC), 118.0 (ArC), 113.8 (ArC), 102.8 (ArC), 68.1 (CH2), 38.7 (CH), 29.3 (OCH3); EI MS peak m/z 439.4 [C24H23O5Cl+] (22%), 383.5 [C21H16O5Cl+] (23%), 257 [C18H9O2+] (28%), 121 [C11H7O3+] (2%), 187 [C11H7O3+] (2%), 183 [C13H11O+] (100%), 93 [C6H5O+] (12%), 71[C4H7O+] (33%), 57 [C3H5O+] (80%), 43 [C3H3O+] (54%).
3-[3-(4-chlorophenyl)-1-(4-fluorophenyl)-3-oxopropyl]-4-hydroxy-2H-chromen-2-one (5k)
Solid; mp 134oC; Yield 72%; ee 99% [S]; IR (acetone) vmax 1720 (C = O), 1610 (C = O), 1490 (C-H), 1300 (O-H); 1H NMR (CDCl3, 200 MHz) δ 9.62 (1H, s, OH), 7.96 (1H, d, J = 1.9 Hz, ArH), 7.93 (2H, d, J = 1.9 Hz, ArH), 7.63 (1H, m, ArH), 7.46 (1H, m, ArH), 7.43 (2H, d, J = 1.9 Hz, ArH), 7.39 (1H, d, J = 2.1 Hz, ArH), 7.29 (1H, m, ArH), 7.15 (1H, dd, J1 = 8.1 Hz, J2 = 1.3 Hz, Ar H), 6.94 (2H, d, ArH, J = 8.6 Hz), 4.82 (1H, d, J = 10.0 Hz, CH2), 4.37 (1H, dd, J1 = 19.1 Hz, J2 = 10.2 Hz, CH2), 4.16 (1H, d, J = 1.6 Hz, CH), 3.68 (1H, s, CH of stereogenic center); 13C NMR (CDCl3, 50 MHz) δ 206.9 (C = O), 167.7 (C = O), 160.9 (C-OH), 152.8 (ArC), 131.8 (ArC-Cl), 130.8 (ArC-F), 130.0 (ArC), 129.8 (ArC), 129.6 (ArC), 129.2 (ArC), 128.8 (ArC), 123.9 (ArC), 116.26 (ArC), 115.18 (ArC), 114.76 (ArC), 68.1 (CH2), 38.7 (CH), 30.9 (CH); EIMS m/z [M+ 1] 422.2 [C24H16O4FCl+] (10%).
Bilogical experiments
Plasma recalcification time (PRT) method.
Anticoagulant potential of test compounds 5a-5k was determined by PRT method.[35] The blood samples were obtained from healthy volunteers in tubes containing 3.8% sodium citrate (9:1) in order to prevent the clotting process. Centrifugation (15 min. at rate 3000 rpm) was carried out to obtain platelet poor plasma. 0.2 ml plasma, 0.1 ml of different concentration of test compounds (100, 300 and 1000 µM) and 0.3 ml of CaCl2 (25 mM) were added together in a clean fusion tube and incubated at 37oC in a water bath. Warfarin was used as positive control. The clotting time was recorded with a stopwatch by tilting the test tubes every 5 sec.
Microplate alamar blue assay (maba) method.
Different derivatives 5a-5k of 4-hydroxycoumarin including warfarin were screened for their antibacterial activities against Escherichia coli and Pseudomonas aeruginosa (gram-negative), Bacillus subtills, Staphyloccus aureus, and Salmonella typhi (gram-positive) by microplate alamar blue assay (maba)[36] using DCM as solvent. The ofloxacin was used as a standard drug. The applied concentration of compounds was 200µg/ml. The zone of inhibition was measured in mm (millimeters) and then %inhibition was calculated.
Agar tube dilution method.
The antifungal activities of all synthesized derivatives 5a-5k of 4-hydroxycoumarin were evaluated with the help of agar tube dilution method.[37] Concentration of samples was 400µg/ml of DMSO Incubated at 37oC and incubation period was 7days. The tested fungal strains were Trichphyton rubrum, Candida albicans, Aspergillus niger, Microsporum canis, Fusarium lini and Canadida glabrata. The Amphotericin B was used as standard drug. The antifungal activities of the compounds were measured in % inhibition.
In silico molecular docking studies
Molecular docking studies were carried out by using Discovery studio 2016, Chemsketch, AutoDock tools-1.5.6 and PyRx. First acquired crystal structure of VKOR1 (PDB ID:3kp9) in PDB format from RCSB Protein data bank,[28] then already attached ligand was removed. Ligands 5a-5k were drawn in chemsketch and assigned smile notation and then open babel was used to add hydrogens and 3D coordinates to convert structures in PDB format. AutoDock tools were used to add polar hydrogens, kollman charges, compute gasteiger charges and set grid box in protein structure and saved it as PDBQT format. Then opened ligand in it and chose torsion for AutoDock using upto 12 torsional degree of freedom (DOF) and then saved it too in PDBQT format. Finally, docking of ligands with VKOR1 was carried out using AutoDock Vina.[38]
{kind=link}
{kind=link}