This study conducted methylation profiling of three HLA-identical T1D families in order to explore epigenetic contribution to the onset of T1D. Exploring methylation differences between affected and unaffected individuals from twins and families minimize variation due to environment or genes and reduce residual noise variance due to factors such as population stratification. An integrative approach involving relevant human transcriptomics data that accelerated the prioritization of candidate genes for T1D was employed.
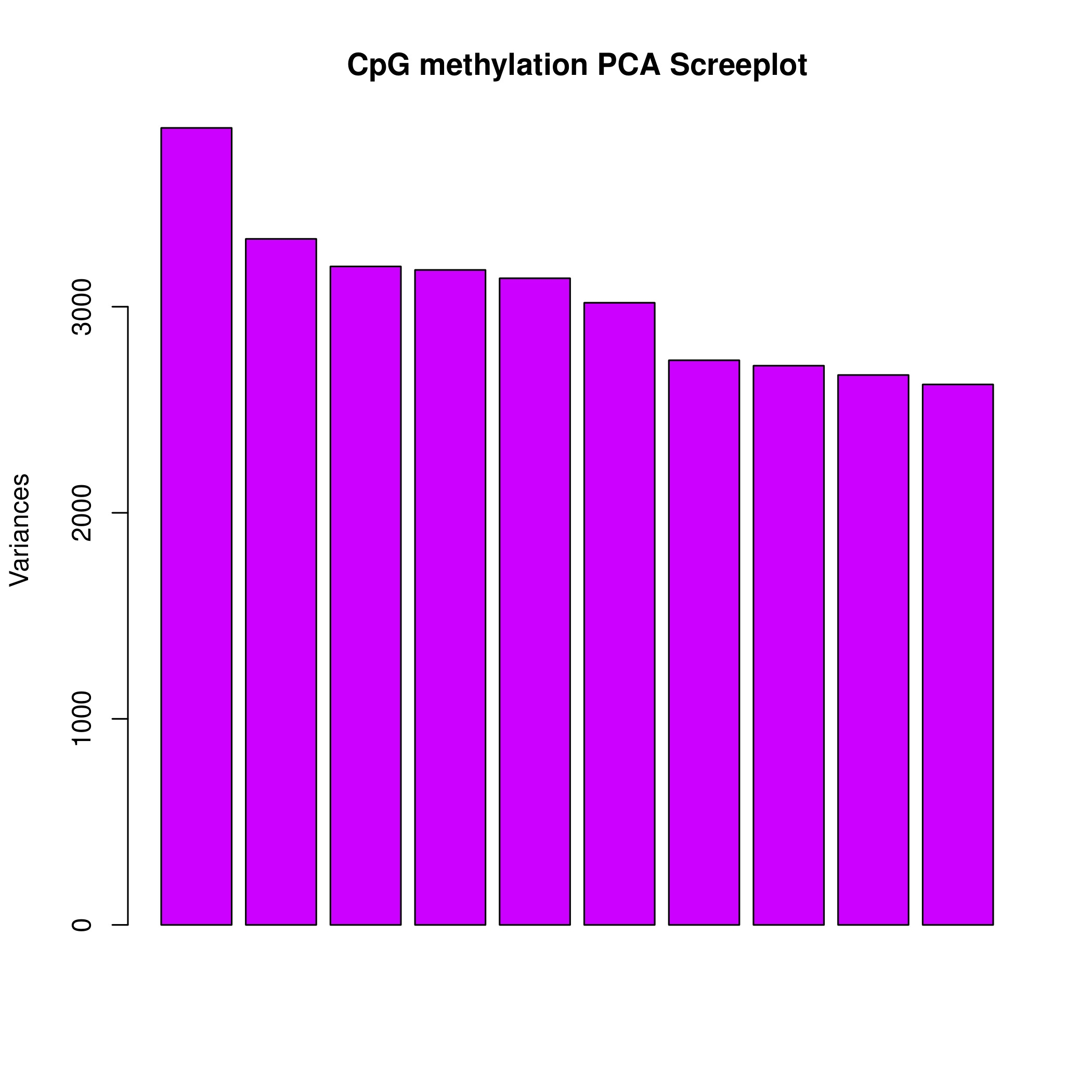
The PCA of methylation profiling of single CpG sites across the three T1D families demonstrated a familywise clustering of affected individuals, which might suggest a common, but family-specific methylation pattern in T1D. Both monozygotic and dizygotic twins showed a tighter cluster in PCA analysis as opposed to that seen with non-twinned siblings, suggesting a similar methylation pattern in twins; thus, such a design is better suited as it helps to reduce spurious differences in methylation events because of confounders. Moreover, a higher degree of closeness could be observed between the monozygotic twins in family B than in the dizygotic twins in family A. This observation adheres to the finding of Hannon et al., (13) who demonstrated that monozygotic twins have higher correlation of site-specific levels of DNAm compared to dizygotic twins.
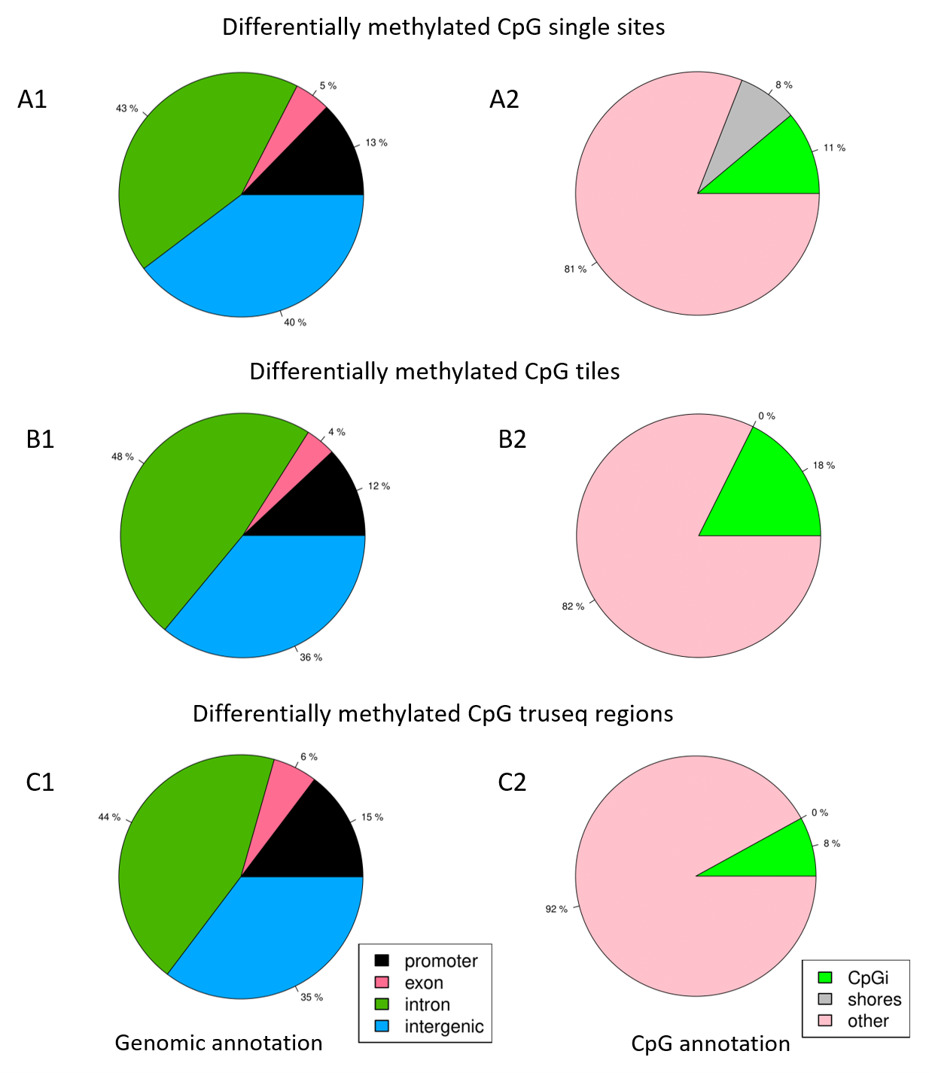
Differential DNAm events were more prominent within intergenic regions and genes, compared to regulatory regions such as promoters, CpG islands, and CpG shores. Similar observations were reported by a study from the Encyclopedia of DNA Elements project that investigated the dynamics of DNAm patterns and relationships to regulatory elements (14). Furthermore, the high percentages of differentially methylated events at promoter regions compared to exons may suggest there is capture enrichment for promoters by the methylation kits target design (15). Although the pathway enrichment analysis revealed the T1D pathway, it was not statistically significant going by FDR value. The inability to identify any significantly enriched pathway is possibly because of the small size of the inputted gene list.
The genomic location of DMC and DMR is crucial in up-/downregulation of gene expression. Moreover, methylation over CpG functional regions (such as CpG island of around 1 Kb in length) - often located near or in the gene promotor region close to the TSS and/or CpG island shore (within 2 Kb from islands) near promoters - may reduce or inhibit gene expression (16). Whereas methylation at sites within a gene, such as the region after the first exon, may result in an increase in gene expression (16).
This study prioritized 18 genes by comparing a set of 84 differentially methylated genes with a published set of differentially expressed genes in T1D. Literature reports support the role of some of these 18 genes in T1D as listed below:
ICA1
This gene is involved in membrane trafficking at the Golgi complex, including transport of insulin in immature secretory granules (17, 18). ICA1 is an autoantigen in insulin-dependent diabetes mellitus (19, 20, 21); however, it has a low number of detectable ICA in Chinese patients compared to Caucasians (22). Expression of ICA1 is high in human pancreatic islets (23) and is upregulated in pancreatic tissue at the clinical onset of T1D (Table 2). The identified hypermethylation events of ICA1 at the gene body correlate with its upregulation expression in our T1D patients (Fig. 3), providing evidence for DNAm mediated regulation of gene expression. However, it is difficult to discern its causality until a fully integrated functional genomics strategy is performed (24). Nevertheless, it is interesting that our study identifies differential methylation events in one of the four major autoantibodies (ICA, GAD, IAA, and IA-2A) that characterize T1D. It is interesting to note that Rakyan et al., (25) observed T1D-associated methylation variable positions (T1D-MVPs) in one of these four autoantibodies, namely GAD2.
MMP26
This gene has been associated with higher fasting plasma glucose levels (26) and impaired wound healing in diabetic patients because of its role in extracellular matrix (ECM) degradation (27).
TIMP3
This gene is functionally related to ECM regulation. Our study identified this gene from both the tiles and targeted DMR analysis. Deficiency of TIMP3 has been associated with insulin resistance and vascular inflammation (28).
DRAM1
We observed hypermethylation of this gene in DMR analysis in the CpGi and promoter regions. Hypermethylation of the CpGi and promoter regions is usually associated with gene silencing. Nevertheless, blood samples from our T1D patients revealed that it was upregulated (Fig. 3). Other research groups observed similar results after performing integrated analysis in which DNAm in functional regions was both concordantly and discordantly associated with gene expression (29, 30). Smith et al., (31) presents that increasing levels of promoter methylation do correlate directly with increased gene expression in a selection of contexts. It is possible to present a context-dependent model whereby epigenetic contributions to transcriptional regulation occur through diverse mechanisms in a more complex and dynamic manner (31, 32). Functionally, the loss of DRAM1 has been demonstrated to inhibit the mechanistic target of rapamycin complex 1 (mTORC1) activation that affects insulin signaling, glycemic balance, and adipocyte differentiation (33).
USP22
This gene suppresses high glucose induced apoptosis in podocytes when silenced (34).
PIK3CG
This gene is known to suppress autoimmune diabetes when suppressed (35).
EXT1
The Exostosin Glycosyltransferase 1 gene encodes an enzyme that is critical for Heparan Sulfate synthesis in β-cells that regulate insulin secretion and glucose homeostasis (36).
The identified DNA methylation events in our study have little in common with those reported in literature. Reasons for this disparity include the following: Apart from one that adapted MZ concordance for T1D (9), reports from literature adapted twins that were T1D-discordant (8, 25, 37, 38). In addition, most of these DNAm studies were array-based which can have an impact on the overall results when compared to NGS-based DNAm profiling. For instance, the methylation profiling studies of Rakyan et al., (25) and Stefan et al., (9) revealed less than 133 CpG sites displaying significant methylation changes using Illumina HumanMethylation 27K array. Disanto et al., (37) and Elboudwarej et al., (38) methylation profiling studies on T1D revealed more than 1,000 CpG sites using Illumina HumanMethylation 450K array. Thus, the results can vary among the same array-based DNAm profiling studies. Moreover, we used HLA-identical (Arab) families, an approach which was used only in one of the above-listed previous studies namely that of Elboudwarej et al., (38) in western population.
The present study has certain limitations. Firstly, we explored specific types of families having mono- or dizygotic twins concordant for T1D phenotype in order to identify novel candidate genes for T1D using NGS-based technology from small size population in Kuwait. This made it difficult to have a large cohort size when compared to other array-based DNA methylation studies in larger population (8, 10). Secondly, blood gene expression profiles from the three families in our study to integrate with DNA methylation events is probably not the best prioritization approach for T1D candidate genes. To compensate, we utilized (a) publicly available relevant transcriptomics data from blood and pancreatic tissue (b) larger cohort to validate the prioritized T1D candidate genes. Thirdly, peripheral blood sample is not the ideal biological sample as (a) it contains a diverse mixture of cells and DNAm is cell-specific (39) (b) it may harbour DNA methylation events due to pathological differences relating to autoimmune processes that we could not statistically adjust for (10). Additionally, Paul et al., (8) demonstrated an increased DNAm variability in T1D across three immune cells. Despite this, Disanto et al., (37) observed a high correlation of methylation events in immune cell types between controls and T1D quadruplets, suggesting a common driving mechanism in more than one immune cell type. Furthermore, blood as biological sample has been used in other DNA methylation studies on T1D (10, 37, 40) as well as in publicly available gene expression data used to prioritize T1D candidate genes in our study. Nevertheless, our future plans would use enriched cell types from the peripheral blood to focus on identifying cell type-specific gene regulatory circuits involved in immune cell metabolism and the cell cycle.
{kind=link}
{kind=link}
{kind=link}
{kind=link}