Materials
Unless stated otherwise, equipment and chemicals used for this study were either products of Thermo Fisher Scientific (Grand Island, NY, USA) or Bio-Rad (Hercules, CA, USA).
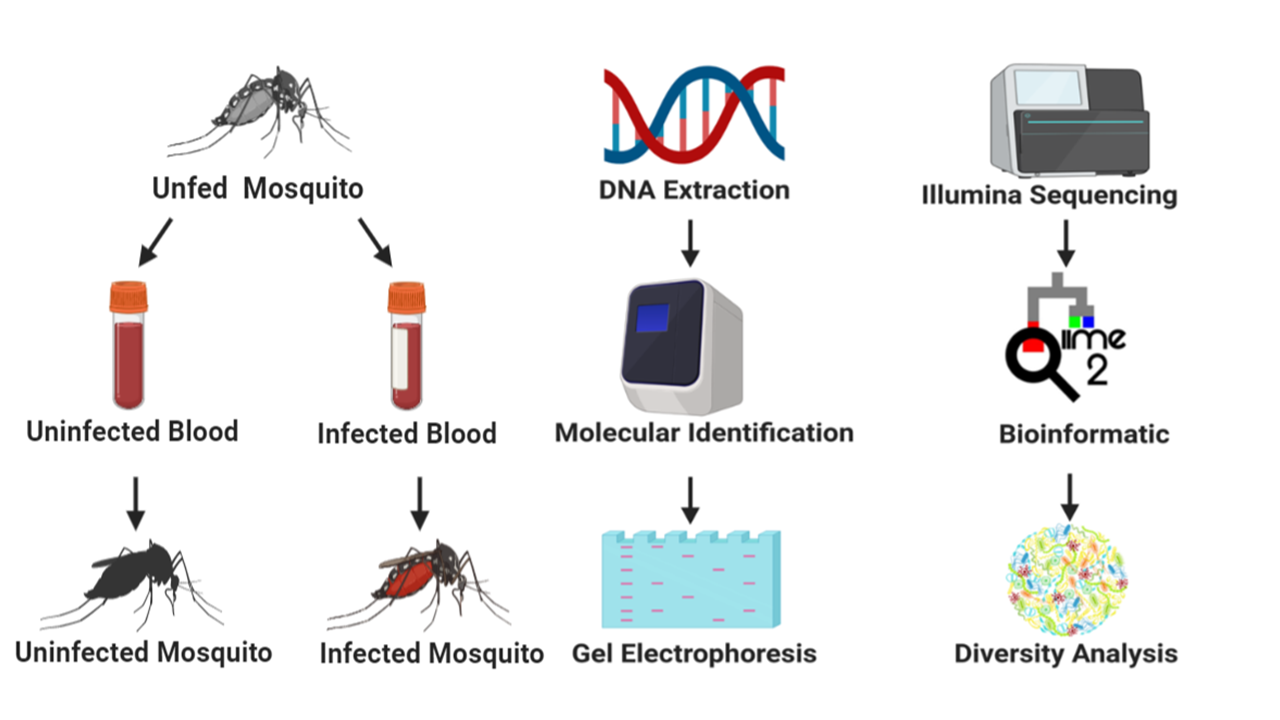
Mosquito rearing and maintenance
The mosquito used for investigation was the Ae. aegypti Liverpool Blackeye strain, a highly susceptible mosquito strain to D. immitis used predominately in research for Aedes spp. [29]. Although Ae. aegypti is just one of 28 mosquito species reported to vector D. immitis in the USA, the relative expertise on this particular species limited us to its use as our study organism. Aedes aegypti, originally obtained from the Filariasis Research Reagent Resource Center (FR3) [30], were raised under standard laboratory conditions: temperature of 27 °C, relative humidity of 80 ± 5%, and a 12:12-hour light:dark diurnal cycle [31].
Mosquito infection with D. immitis
Adult female mosquitoes, five days post-emergence were blood-fed using an artificial membrane feeder. One day prior to membrane feeding 31 female mosquitoes were transferred to ~500 ml plastic containers with mesh tops (henceforth “cages”). Females were starved of sugar for 12 h and deprived of water for 4 h prior to blood feeding. Mosquitoes in each cage (31 each) were allowed to feed for 2 h or until repletion on a Parafilm membrane stretched over an inverted water-jacketed glass membrane feeder maintained at 40 °C. Each feeder was filled with 200 µl of dog blood infected or uninfected with D. immitis. The level of parasitaemia in the infected blood was estimated to be 4500 mf/ml as previously reported [31]. The dog blood was obtained from FR3.
PCR-based confirmation of Dirofilaria immitisdetection
Prior to screening for D. immitis infection from mosquitoes, DNA from individual mosquito samples was extracted using a DNeasy Blood & Tissue Kit (Qiagen, Germantown, Maryland, USA) and quality was confirmed using a nanodrop machine (Nanodrop One, Thermo Fisher Scientific). All blood-fed (infected and uninfected blood) mosquitoes were screened for D. immitis infection irrespective of whether they were fed on infected or uninfected blood by amplifying the cox1 gene (656 bp) of the D. immitis mitochondrial DNA [32]. Briefly, a 25 µl reaction was set up comprising 1 µl each of the COI-Forward (5'-TGA TTG GTG GTT TTG GTA A-3') and COI-Reverse (5'-ATA AGT ACG AGT ATC AAT ATC-3') primers, 12.5 µl of 2× mastermix (New England Biolabs, Ipswich, Massachusetts, USA), 2.5 µl DNA template and 8.5 µl of nuclease-free water. For each cycle that was run, a D. immitis infected blood sample and nuclease-free water were simultaneously included as positive and negative controls respectively. The PCR cycle comprised of an initial denaturation step at 94 °C for 5 min and 40 cycles of 94 °C for 1 min, annealing at 50 °C for 2 min and extension at 72 °C for 3 min followed by a final extension step at 72 °C for 5 min and an infinite hold at 4 °C.
Confirmation of amplification was done by loading the PCR products in a SYBR safe stained gel. Briefly, 2% gel was made by autoclaving a solution of 1× TAE buffer and molecular grade agar. SYBR safe stain (1 µl SYBR safe: 10 ml TAE buffer) was added to the agar solution, poured into a precast gel tray and allowed to cool. To load the samples onto the gel, 6 µl of PCR product was mixed with 4 µl of 6× dye and pipetted into the wells. Lastly, 5 µl of a low molecular weight DNA ladder was loaded onto the gel and the gel could run for 45 min at 100 V. Amplified PCR products were viewed using a Chemidoc gel imager (Additional file 1: Figure S1).
16S rRNA library preparation and sequencing
Six individual mosquito genomic DNA extracts were pooled to make one biological replicate and five biological replicates each of D. immitis infected and uninfected pools were prepared for metagenomic analysis. The hypervariable V1-V3 region of the 16S rRNA gene was PCR amplified using the forward primer 27F (5'-AGR GTT TGA TCM TGG CTC AG-3') and the reverse primer 519R (5'-GTN TTA CNG CGG CKG CTG-3') as outlined by the 16S Illumina’s MiSeq protocol (www.mrdnalab.com, Shallowater, TX, USA). Briefly, PCR was performed using the HotStarTaq Plus Master Mix Kit (Qiagen) under the following conditions: 94 °C for 3 min, followed by 30–35 cycles of 94 °C for 30 s, 53 °C for 40 s and 72 °C for 1 min, after which a final elongation step at 72 °C for 5 min was performed. After amplification, PCR products were electrophoresed in 2% agarose gel to determine the success of amplification and the relative intensity of bands. Multiple samples were pooled together in equal proportions based on their molecular weight and DNA concentrations. Pooled samples were purified using calibrated Ampure XP beads. Then the pooled and purified PCR product was used to prepare Illumina DNA library. Sequencing was performed at MR DNA (www.mrdnalab.com, Shallowater, TX, USA) on a MiSeq following the manufacturer’s guidelines.
Sequence analysis
Sequence analysis was carried out using the Quantitative Insights into Microbial Ecology (QIIME 2) pipeline, unless stated otherwise. Briefly, processing of raw fastq files were demultiplexed. The Atacama soil microbiome pipeline was incorporated for quality control of demultiplexed paired-end reads (Additional file 1: Figure S2) using the DADA2 plugin as previously described [33].
Sequence alignment and subsequent construction of phylogenetic tree from representative sequences was performed using the MAFFT v7 and FasTree v2.1 plugin [34] Operational taxonomic assignment was performed using the qiime2 feature-classifier plugin v7.0 which was previously trained against the SILVA 132 database preclustered at 99%. Tables representing operational taxonomic units (OTUs) and representative taxonomy were exported from R and used for diversity metric analysis using the Microbiome Analyst web-based interface [35, 36]. Raw data from this analysis were deposited and assigned the GenBank BioProject number #PRJNA606536.
Alpha diversity
To establish whether alpha diversity differs across mosquito samples, reads were transformed and low abundance OTUs were filtered from the datasets. The Observed OTU metric was used to estimate species richness by identifying unique OTUs present across the mosquito groups, while the Shannon index was used to estimate both richness and evenness.
Beta diversity
To compare the differences in the microbiome between mosquito groups based on measures of distance or dissimilarity, dissimilarity matrix was generated from log-transformed sequence data and ordination of the plots were visualized using both the principal coordinates analysis (PCoA) and the non-metric multidimensional scaling (NMDS). The matrix used in calculating beta diversity includes the Bray-Curtis and unweighted UniFrac distance matrix.
Statistical analysis
To test if species richness and diversity was significant, the Mann-Whitney or Kruskal-Wallis tests was applied to both alpha diversity and classical univariate statistical comparisons analysis, while the significance of beta diversity analysis was determined using the permutational MANOVA (PERMANOVA) test [35, 36].
{kind=link}