Materials
Far, farnesol, and TBAP were supplied by Sigma-Aldrich (St. Louis, MO, USA). Chlorhexidine digluconate (CHX) was obtained from Macklin (Shanghai, China). HO-PEG2000-NHNH2 was obtained from Xi'an Ruixi Biological Technology (Xi'an, China), and mPEG2000-PLA2000 was purchased from Jinan Daigang Biomaterials (Jinan, China). Mitis Salivarius agar was purchased from BD Biosciences (San Jose, CA, USA). Brain heart infusion broth and agar were purchased from Hopebio (Qingdao, China). S. mutans UA159 (ATCC 700610) was purchased from the China Center of Industrial Culture Collection (Beijing, China).
Synthesis of pH-sensitive dentotropic polymeric conjugate
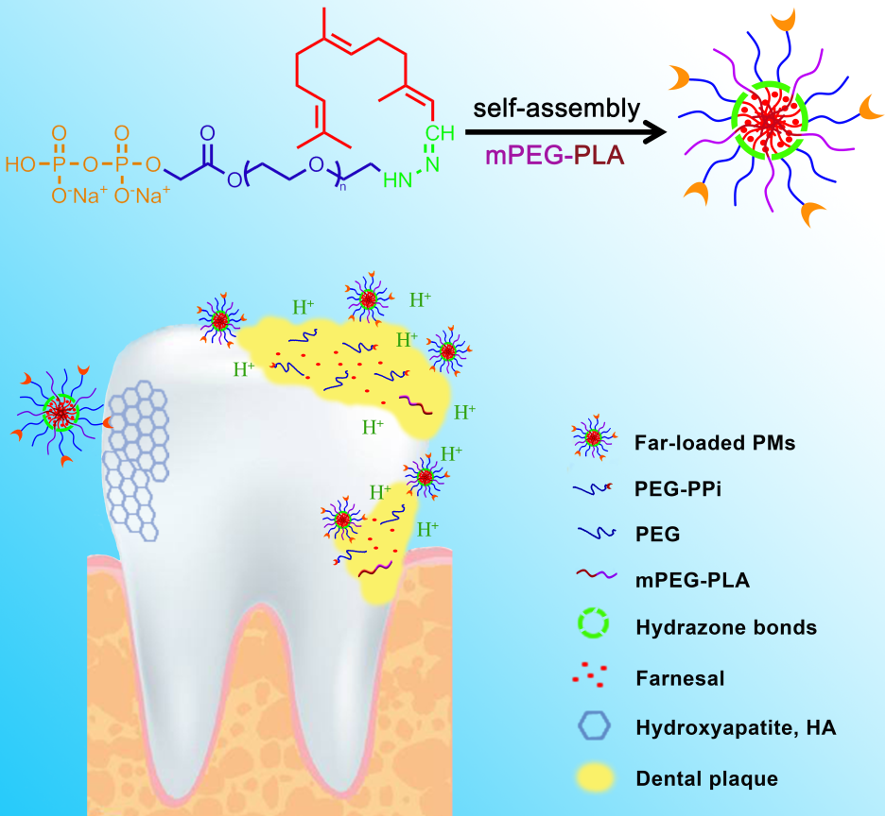
The pH-sensitive dentotropic polymeric conjugate PPi-PEG-hyd-Far was synthesized as shown in Figure 1. The commercially available HO-PEG2000-NHNH2 was protected with a t-butyloxy carbonyl (Boc) group, and then subjected to esterification and bromination to give PPi-PEG-NHNH-Boc. The Boc group was removed and subsequent hydrazide reaction generated PPi-PEG-hyd-Far, whose identity was confirmed using 1H and 13C NMR. The specific reaction conditions for each compound are detailed below.
Synthesis of compound 1: tert-butyl 2-(2-(2-hydroxyethoxy)acetyl)hydrazine-1-carboxylate (HO-PEG-NHNH-Boc)
Triethylamine (25.3 mg, 0.25 mmol) was added to a stirred solution of HO-PEG2000-NHNH2 (500 mg, 0.25 mmol) in methanol. The mixture was cooled to 0 °C and di-tert-butyl dicarbonate (65.5 mg, 0.30 mmol) was added slowly to the reaction solution. The reaction mixture was stirred at 0 °C for 1 h, then warmed to room temperature and stirred for 24 h. The solution was concentrated under vacuum, and the product was precipitated in anhydrous ether three times, dried overnight under vacuum and stored at -20 °C for an overall yield of 72.4%.
Synthesis of compound 2: sodium 2,2-dimethyl-4,7,13-trioxo-3,9,12-trioxa-5,6-diazatetradecan-14-yl hydrogen diphosphate (PPi-PEG-NHNH-Boc)
Bromoacetic acid (43.4 mg, 0.31 mmol) was added to a stirred solution of compound 1 (500 mg, 0.24 mmol) in anhydrous dichloromethane (DCM). The solution was cooled to 0 °C, then 4-dimethylaminopyridine (DMAP, 2.9 mg, 0.024 mmol) and N,N’-dicyclohexylcarbodiimide (DCC, 54.4 mg, 0.26 mmol) were added slowly, and the mixture was stirred at room temperature for 24 h. The reaction mixture was filtered and concentrated under vacuum, and the product was precipitated in anhydrous ether. The precipitate was filtered and dialyzed against water for 24 h. TBAP (415.1 mg, 0.46 mmol), previously dissolved in anhydrous acetonitrile (CH3CN), was added slowly to the dialyzed product, and the mixture was stirred at room temperature for 12 h. The solution was concentrated under vacuum, and the product was precipitated into anhydrous ether. The product was filtered, dialyzed against NaCl solution and then dialyzed against water at 0 °C for 10 h. The purified product was freeze-dried and stored at -20 °C for overall yield of 80.7%.
Synthesis of compound 3: sodium 2-(2-(2-hydrazinyl-2-oxoethoxy)ethoxy)-2-oxoethyl hydrogen diphosphate (PPi-PEG-NHNH2)
Compound 2 (500 mg, 0.18 mmol) was de-protected using zinc bromide (ZnBr2, 81 mg, 0.36 mmol) in dichloromethane (DCM) for 4 h . The solution was filtered and concentrated under vacuum, and the product was precipitated in anhydrous ether three times and against water at 0 °C for 10 h.. Then the purified product was dried overnight under vacuum and stored at -20 °C with overall yield of 80.20%.
Synthesis of compound 4: sodium 2-oxo-2-(2-(2-oxo-2-(2-((2E,6E)-3,7,11-trimethyldodeca-2,6,10-trien-1-ylidene)hydrazinyl)ethoxy)ethoxy)ethyl hydrogen diphosphate (PPi-PEG-hyd-Far)
Compound 3 (500 mg, 0.19 mmol) and Far (61.0 mg, 0.28 mmol) were dissolved in anhydrous methanol, and acetic acid (1.1 mg, 0.02 mmol) was added to the reaction solution. After stirring at room temperature for 48 h, the solution was concentrated under vacuum, and the product was precipitated in anhydrous ether three times. Then the purified product was dried overnight under vacuum and stored at -20 °C with overall yield of 79.80%.
Preparation of polymeric micelles
The membrane hydration method [37] was used to prepare blank polymeric micelles (PMs), Far-loaded pH-sensitive polymeric micelles (Far-PMs), as well as PPi-targeted and Far-loaded pH-sensitive dentotropic polymeric micelles (PPi-Far-PMs). The following components were used: mPEG2000-PLA2000 (A), PEG-hyd-Far (B), PPi-PEG-hyd-Far (C), and Far (D). The weight ratios of components A/B/C/D were optimized using the central composite design [38-39] to be as following (Table 1): blank PMs, 40/0/0/0; Far-PMs, 20/20/0/4; and PPi-Far-PMs, 20/0/20/4. The components were dissolved in 4.0 mL of acetonitrile in a round-bottom flask, then the acetonitrile was evaporated under vacuum at 55 °C to obtain a thin film. Residual acetonitrile was completely removed under vacuum overnight at room temperature. The dried thin film was hydrated with ultra-pure water (2.0 mL) and sonicated for 3 min in a bath sonicator, stirred for 12 h at room temperature and filtered through a 0.22-µm membrane. The micellar solution was freeze-dried and stored at 4 °C.
The same method and the additional reagent FITC-PEG (E) were used to prepare FITC-labeled PMs in the following weight ratios: Far-PMs-FITC, A/B/D/E: 20/20/4/1; and PPi-Far-PMs-FITC, A/C/D/E: 20/20/4/1.
Characterization of polymeric micelles
The size and zeta potential of polymeric micelles were measured using dynamic light scattering (Zetasizer Nano ZS90, Malvern Instruments, Malvern, UK). Morphology of blank PMs and PPi-Far-PMs were observed using transmission electron microscopy (JEM-100SX, Japan). Critical micelle concentration (CMC) was determined using fluorescence spectroscopy and pyrene as the hydrophobic fluorescent probe.
Encapsulation and drug-loading efficiencies were calculated using the ultrafiltration method and an ultrafiltration column with a molecular weight cut-off of 3 kDa (Solarbio Science and Technology, Beijing, China). Far was quantified on an Agilent ZORBAXSB-C18 column (4.6 × 150 mm, 5 µm) at 25 °C attached to an Agilent 1260 HPLC system (Infinity, USA). The mobile phase was acetonitrile and ultrapure water (80:20, v/v) and the flow rate was 1 mL/min. The detection wavelength was 216 nm. Encapsulation efficiency was calculated using the equation:
Encapsulation efficiency (%) = (Weight of Far in micelles) / (Weight of total Far) × 100%.
Drug-loading efficiency was calculated using the equation:
Drug-loading efficiency (%) = (Weight of Far in micelles) / (Weight of total micelles) × 100%.
Release of Far from PPi-Far-PMs was investigated in vitro using the dialysis method. Briefly, 1 mL of PPi-Far-PMs or free Far were placed into separate dialysis bags with a molecular weight cut-off of 1 kDa (Spectrumlabs, USA) and dialyzed at 37 °C against phosphate-buffered saline (PBS) at pH 4.5 and 7.4 with gentle stirring. At different time points (0.1, 0.2, 0.4, 0.5, 1, 2, 4, 6, 8, 12 and 24 h), 0.5 mL of release medium was removed and replaced with fresh medium. Far was quantified by HPLC as described above, and experiments were performed three times.
Binding of PPi-Far-PMs to hydroxyapatite particles in vitro
As we all know, the most important component of the tooth enamel is hydroxyapatite [24]. To mimic the binding process of PPi-Far-PMs with tooth enamel, we used a small intestinal submucosa as the bio-mineralization template to prepare plate-like, single-crystal hydroxyapatite, referred to henceforth as ‘biotechnological hydroxyapatite’. Meanwhile, we used commercially available hydroxyapatite (Macklin, Shanghai, China) as the control.
Synthesis and characterization of hydroxyapatite
Small intestinal submucosa was prepared as described [40]. The reaction device included a beaker and a centrifuge tube with a hole in the middle of the cap. The submucosa membrane covered the hole to seal the centrifuge tube, which was filled with a solution of K2HPO4 (30 mL, 0.1 M). This tube was inverted and soaked into the beaker filled with a solution of Ca(CH3COO)2 (30 mL, 0.1 M). This reaction system, which mimics bone mineralization conditions, was incubated at 37 °C for 10 days. Care was taken to ensure that the top and bottom surfaces of the submucosa membrane remained in contact with liquid [41]. Morphology of the biotechnological hydroxyapatite was analyzed using scanning electron microscopy (Inspect F50, FEI, America).
Binding potential and kinetics of PPi-Far-PMs on hydroxyapatite
Solutions of FITC-labeled PPi-Far-PMs and Far-PMs were mixed with biotechnological or commercial hydroxyapatite in round-bottom flasks (at pH 7.4). After incubation with gentle stirring for 30 min at room temperature, the mixture was filtered, washed with PBS three times, and freeze-dried. Finally, the hydroxyapatite was observed under a fluorescence microscope (FL, AMG, America).
For analysis of binding kinetics, PPi-Far-PMs or Far-PMs (1 mL) were mixed with either kind of hydroxyapatite particles (50 mg) and incubated at room temperature. At certain time points (0, 0.5, 1, 5, 30 and 720 min), hydroxyapatite was removed by centrifugation (10,000 g, 5 min) and the supernatant was collected. The amount of Far in the micelle supernatant after binding (Wleft) was analyzed by HPLC as described above. The binding rate (%) at each time point was calculated as follows: Binding rate (%) = (Wtotal – Wleft) / Wtotal × 100%.
Anti-bacterial activity of Far
The ability of Far to kill S. mutans UA159, a proven virulent cariogenic dental pathogen, was assayed. S. mutans was stored at -80 °C as stocks in brain heart infusion broth containing 25% (v/v) glycerol. These stocks were streaked onto Mitis Salivarius agar and incubated anaerobically for 48 h at 37 °C under an atmosphere of 80% N2, 10% H2 and 10% CO2. Then a single colony of bacteria was inoculated into brain heart infusion broth, and the culture was incubated anaerobically for 12 h at 37 °C. Bacterial growth was assayed by measurement of absorbance at 600 nm. Optical densities were converted to CFU/mL using the conversion (0.64 = 1 × 109 CFU/mL [10]).
Minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) of Far were determined against S. mutans using broth microdilution [42]. The initial inoculum was 5 × 105 CFU/mL, and the concentration of Far ranged from 3.5 to 448 μg/mL with two-fold dilutions. Farnesol and chlorhexidine digluconate were assayed in parallel as controls. MIC was defined as the lowest concentration that showed no growth in the medium after anaerobic incubation for 48 h. Brain heart infusion broth on its own was assayed as a blank control, while the same medium supplemented with DMSO (0.076%, v/v) was assayed as a solvent control. MBC was defined as the lowest concentration that showed no surviving bacteria on the agar after incubation for 48 h at 37 °C. For MBC determination, medium in micropores without bacterial growth was picked up with a sterile inoculating loop and plated onto brain heart infusion blood agar supplemented with 5% defibrillated sheep blood. These experiments were performed three times.
In vivo anti-caries efficacy of PPi-Far-PMs
Sprague-Dawley rats were obtained from the Laboratory Animal Center of Southwest Medical University (Luzhou China). All animal experiments were approved by the Animal Ethics Committee of this university (permit 2017060012) and carried out in accordance with the Luzhou municipal government guidelines on animal care and use.
To establish a rat model of dental caries, animals were fed a cariogenic diet with 56% sucrose (Keyes 2000; Beijing Keao Xieli Feed, Beijing China) and given 5% sucrose water to drink ad libitum [1, 43]. After weaning, 17-day-old Sprague-Dawley rats (specific pathogen-free) were fed with sodium ampicillin (0.1% in food) for 4 days to inhibit endogenous bacterial growth in the oral cavity. Any animals infected with S. mutans prior to inoculation were removed from the study on day 21. Then each rat was inoculated with 1 mL S. mutans (7×108 CFU/mL)every day for 7 days. After being checked for infection, 29-day-old rats were randomly divided into nine groups (n=7), and their teeth were topically treated twice daily for 5 weeks with a camel hair brush coated with the following treatments: (a) distilled water (negative control), (b) 15% ethanol (v/v, vehicle control), (c) farnesol (1.10 mg/mL), (d) farnesal (Far; 1.10 mg/mL), (e) chlorhexidine gluconate (1.10 mg/mL; positive control), (f) blank PMs, (g) Far-PMs (1.10 mg/mL Far) and (h) PPi-Far-PMs (1.10 mg/mL Far), ensuring that the drug kept working with the teeth for 1 min. All animals were weighed weekly, and physical appearance was recorded daily. At the end of the 5 weeks, saliva was collected and inoculated onto Mitis Salivarius agar with bacitracin (Sigma) to estimate the S. mutans population, and on brain heart infusion agar with 5% sheep blood to determine the total colony count. Finally, animals were sacrificed and the teeth were collected for further assessment.
To evaluate the anti-caries activity of PPi-Far-PMs, teeth were stained with 0.4% murexide solution, and caries were scored according to Keyes’ system [36]. A stereomicroscope (M205FA, Leica, Germany) was used to assess caries severity on the smooth-surface (Smo), proximal-surface (Pro), and sulcal-surface (Sul). Severity was graded on a four-level scale: enamel affected (E), dentin exposed (Ds), 3/4 of the dentin affected (Dm) and all dentin affected (Dx). Classifications in this study are written in the form "surface-severity", e.g. Smo-E or Sul-Dx. The extent of caries reduction was determined using the following formula:
Caries reduction (%) = (Keyes’s score of negative control group - Keyes’s score of test group) / (Keyes’s score of negative control group) × 100%.
Rat teeth were collected and assessed in terms of mechanical characteristics and microarchitecture. Compressive strength of teeth was measured using a universal testing machine (Meister E44, USA) at 37 ± 0.5 °C. A stress-strain curve was recorded as vertical tooth occlusal pressure was applied at 1 mm/min. After the samples were broken, the compression modulus (slope of stress-strain curve), compressive strength, compressive yield stress, and maximum stress (the highest point of stress-strain curve) were calculated base on the stress-strain curve. Microarchitecture of molars was evaluated using high-resolution micro-computed tomography (micro-CT; skyscan1172, Bruker Corporation). The following parameters were used: voltage, 80 kV; current, 80 μA; exposure time, 2.96 s; scan resolution, 14 μm/slice; and total rotation angle, 360° increasing in 0.5° increments. Bone mineral density (BMD) and bone volume per tissue volume (BV/TV) were estimated from the three-dimensional reconstructions.
Statistical analysis
All data were expressed as means ± standard deviation (SD) and analyzed using GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA, USA). Differences between treatment groups were analyzed for significance using the Student’s t test or Tamhane's T2 test. P < 0.05 was regarded as statistically significant.
{kind=link}