Docking study
The assessment of the biological activity of the proposed structures as well as the preselection of molecules for synthesis was performed using AutoDock Vina20 and AutoDock4.2 software packages21,23. As mentioned above, 2-pyridones with potential anticancer activity often act on PI3K, which are responsible for cellular growth and survival signals and thus regulate tumor growth and expansion. Therefore, for the evaluation of our molecules, we chose PI3K from the PI3K/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) pathway. In silico calculation was performed using the crystallographic structure of two kinase isoforms— human γ24 and mouse δ25 PI3K—obtained from PDB. We used the PI3K δ kinase complexed with Leniolisib, a well knowns and selective PI3K δ inhibitor, to verify if Leniolosib will dock at the same position as was observed in the crystallographic structure. The analysis showed a positive result, and docked Leniolisib overlapped the molecule present in the crystallographic structure, with a binding energy value of –8.84 kcal/mol and inhibition constant value of 333.92 nM. Thus, we carried out a docking study with our designed ligands. In the first attempt, we docked a series of compounds (20) to PI3K δ. Lock-through of the estimated binding energy values pointed out that the highest affinity to PI3K δ was shown by the molecules with N-aryl urethane moiety (20). The lowest binding energy (–10.11 kcal/mol), as well as inhibition constant value (38.9 nM) was observed for (R)-20d. (R)-20d exhibited hydrophobic interactions with TRP 760, ILE 777, TYR 813, ILE 825, PHE 908, ILE 910, and ASP 911. Hydrogen bond formation was observed between the urethane moiety of (S)-20d and VAL 828 and SER 831 (Figure 3a).
Next, we evaluated a series of chloropyridine derivatives (18) to determine their binding mode. Even in this case, N-aryl urethanes exhibited the highest affinity to kinase. Compound (S)-18d had the lowest binding energy in this series (–10.88 kcal/mol) and a slightly higher inhibition constant (10.6 nM) than 2-pyridone derivative. The binding mode of (S)-18d is presented in Figure 3b. The compound showed hydrophobic interactions with TYR 813, ILE 825, PHE 908, and ILE 910. In addition, hydrogen bond formation was observed between the urethane moiety of (S)-18d and VAL 828, and π-stacking between TRP 760 and 3-fluorophenyl ring. We also tested the binding mode of a series of methoxypyridine derivatives (19). Interesting docking results were observed for (R)-19d (Figure 3c) with a binding energy value of –10.53 kcal/mol and inhibition constant value of 19.1 nM. Similar hydrophobic interactions were observed between TYR 813, ILE 825, and ILE 910, and additional interaction between ILE 777 with a cyclohexyl ring and ASP 911 with a phenyl ring. Urethane formed hydrogen bond with VAL 828 and additionally with SER 831. In addition, π-stacking was observed between TRP 760 and 3-fluorophenyl ring.
In the next step, we checked the modes of binding of our designed ligands to PI3K γ. We noted that interaction with protein was not as effective as that observed for PI3K δ; however, the binding energy calculated for (S)-20d (binding energy: –10.08 kcal/mol, inhibition constant: 40.7 nM) was better than that of Leniolisib. A similar pattern of interactions was observed: hydrophobic interactions with ILE 831, TYR 867, ILE 879, ILE 881, ILE 963, and ASP 964; hydrogen bond between the urethane moiety of (S)-20d and VAL 882, and a π-stacking interaction between TRP 812 and 3-fluorophenyl ring, as well as between TYR 867 and 2-pyridone ring (Figure 3d). Summing up, our in silico calculation confirmed that our tetrahydroquinolin-2(1H)-ones showed a consistent mode of interaction with kinase. The following parts of the ligand were of key importance in the interactions: 3-fluorphenyl ring for π-stacking, urethane for hydrogen bond with VAL 828, and bicyclic tetrahydroquinoline moiety for hydrophobic interaction. Moreover, docking showed that all considered structures exhibited a higher affinity to the active site of PI3K δ than for the reference compound Leniolisib. These results prompted us to synthesize a series of new tetrahydroquinolin-2(1H)-ones and evaluate their biological activity.
Chemistry
Synthesis of tetrahydroquinoline derivatives
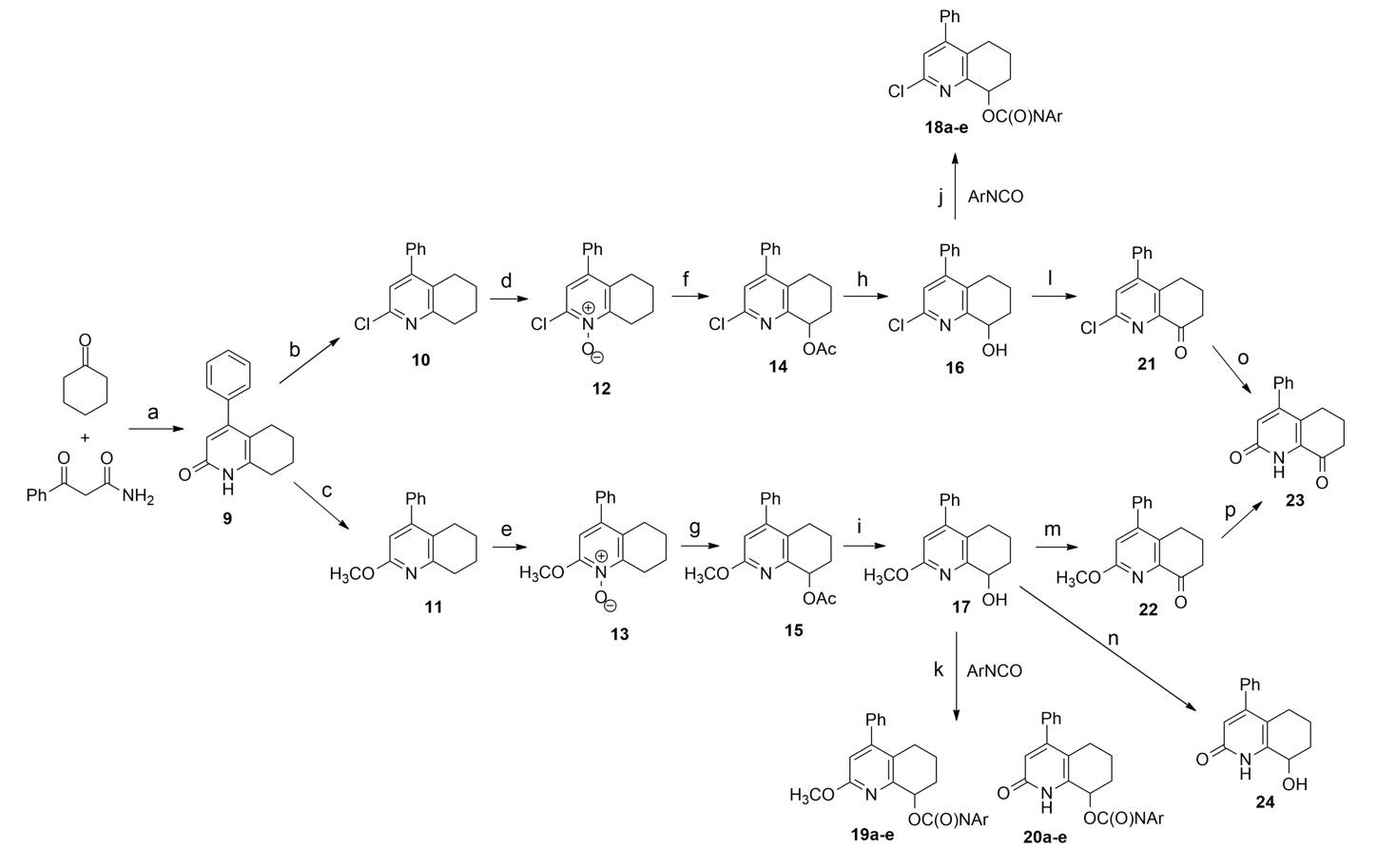
We synthesized the new PI3K inhibitors through the following pathway (Scheme 1). In the first step, we used a modified procedure for the synthesis of tetrahydroquinolin-2(1H)-ones26,27 and tetrahydroquinolin-2(1H)-one core (9). During condensation, we found out that it is additionally necessary to remove water formed with anhydrous MgSO4. Since it was not possible to perform direct functionalization of 9 at position 8, we had to transform tetrahydroquinolin-2(1H)-one (9) into 2-chlorotetrahydroquinoline (10) and 2-methoxytetrahydroquinoline (11) via typical chlorination with PhP(O)Cl228 and alkylation with silver carbonate29. Compounds 10 and 11 were oxidized at position 8. However, this two-step process required the formation of N-oxides (12, 13) which subsequently underwent intramolecular disproportionation into 8-acetoxy derivatives (14, 15). The acetyl group was removed by alkaline hydrolysis. Structures 16 and 17 were important for multiway functionalization. The first pathway led to the formation of 2-chloro-4-phenyl-5,6,7,8-tetrahydroquinolin-8-yl arylcarbamates (18a–e) by the addition of alcohol 16 to various isocyanates in the presence of tertiary amine. Formation of 6,7-dihydroquinolin-8(5H)-one derivatives (21, 22) required to perform Swern oxidation. The compound 4-phenyl-6,7-dihydroquinoline-2,8(1H,5H)-dione (23) might be independently formed through acidic hydrolysis of 21 or hydrolysis of 22 with acetic acid catalyzed by NaI. It must be noted that 8-hydroxytetrahydroquinolin-2(1H)-one (24) can only be formed from 17 via hydrolysis with AcOH and NaI, because acid hydrolysis of 2-chloro-tetrahydroquinolin-8-ol (16) led to the formation of a dehydrated product. However, the results of our subsequent experiments indicated that the preparation of intermediate 24 was not necessary at all. From the reaction mixtures consisting of 17, triethyl amine, and appropriate isocyanate, we isolated a carbamate derivative of the parent compound (19) together with a significant amount of 2-oxo-4-phenyl-1,2,5,6,7,8-hexahydroquinolin-8-yl arylcarbamate (20). Generally, the amount of secondary products formed depends on the used isocyanate; however, we observed the aforementioned phenomenon. We experimentally evaluated several possible ways for demethylation.
To track the migration of the methyl group, we prepared 17 with a 13C-labeled methyl group. We introduced the labeled methyl group using commercially available 13CD3I as an alkylating agent. Surprisingly, our attempts to perform an identical reaction between 13CD3-labeled 17 and p-nitrophenyl isocyanate in the presence of amine were not successful. 13CD3-labeled 17 does not undergo demethylation. The first obvious solution was to remove the methyl group by a nucleophilic attack, for example, using water. However, during nuclear magnetic resonance-monitored reaction with nonlabeled 17, we did not observe any CH3 signal of methanol in the reaction mixture using a pyridine base to uncover the aliphatic part of the spectrum. The thin-layer chromatographic analysis of this reaction mixture revealed the formation of the demethylated product 20. We also observed that strong exclusion of water from the reaction mixture during the process may inhibit demethylation, which may be due to, for example, the molecular sieves present in the solvent. Therefore, we postulated that demethylation is caused by traces of water. Additionally, we found that compound 19, which was formed already, did not undergo demethylation, even if it was treated with a fresh portion of isocyanate. Similarly, pure 17 was not demethylated in solution in the absence of isocyanate.
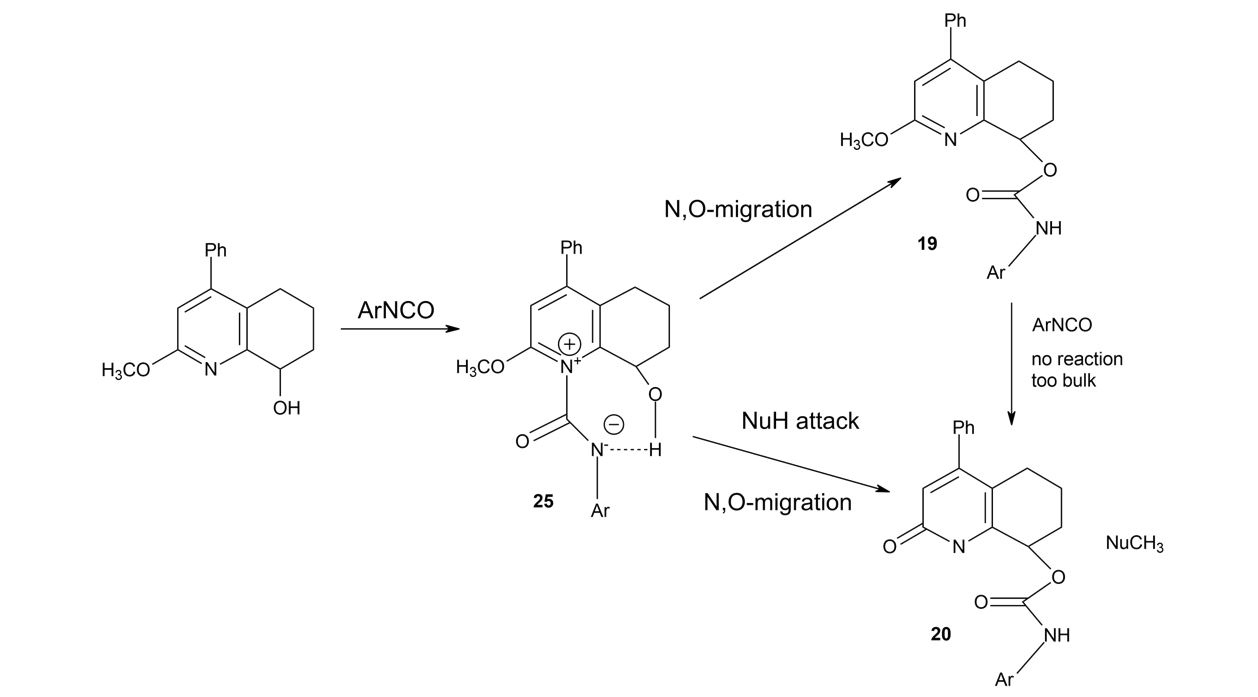
Nevertheless, although the mechanism of demethylation was still unclear, we assumed a tentative reaction mechanism to elucidate the observed process (Scheme 2).
At the first step, 2-methoxy-4-phenyl-5,6,7,8-tetrahydroquinolin-8-ol (17) was acylated with isocyanate on the nitrogen atom. The resulting intermediate 25 was susceptible to nucleophilic attack on the methyl group due to the positive charge of nitrogen. In the next step, the migration of isocyanate moiety from pyridinium nitrogen to alcoholic oxygen was carried out in order to terminate the process. This step might be accompanied by simultaneous nucleophilic attack of water on the methyl group, leading to the formation of demethylation product 20. The already formed product 19 did not undergo demethylation because nitrogen atom is associated with steric hindrance, which excludes the possibility of its reaction with isocyanate.
Thus, the complete lack of demethylation in experiments with 13CD3-labeled 17 strongly suggests the existence of reversed secondary isotope effects, which can influence the reaction rate, and indicates that the observed demethylation reaction should be of Sn2 type or a similar type involving modified sp3/sp2 hybridization of the methyl group.
Biological activity
Tetrahydroquinoline derivatives displays potent cytotoxic and antiproliferative activities
To evaluate the effects of the newly synthesized tetrahydroquinolin-2(1H)-one derivatives on cell viability, we treated human colon cancer cells HCT-116, human breast cancer cells MCF-7, and human non-small cell lung cancer cells A-549 with different concentrations (0 and 50 μM) of the compounds for 72 h. After incubation, the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was performed and the cytotoxic potency of compounds (expressed in IC50) on the cells was determined in comparison to control cells treated with 1% of dimethyl sulfoxide (DMSO) (Table ). Compounds 9, 16, 18b, 18d, 21, 23, 22, 17, 24, and 20e showed no cytotoxicity on tested cell lines at concentrations up to 50 µM. Other compounds showed moderate activity against A-549 and HCT-116 cell lines, while none of the compounds displayed cytotoxic effect on MCF-7 breast cancer cells. The IC50 values were estimated at the lowest micromolar concentration (cytotoxicity at ~13 µM) for compounds 19b, 19c, 19e, 20a, and 20d in HCT-116 cells among the studied derivatives. While these derivatives showed pronounced cytotoxicity against colon cancer cells, only compounds 20d and 19b exhibited similar potency against A-549 lung cancer cells (Figure 4a and Figure 4b). We also compared the activity of 20d and 19b to the reference control drug Cisplatin, which is widely used in chemotherapy30. It was observed that the tested compounds were more effective than Cisplatin in both HCT-116 and A-549 cell lines (Table 1), suppressing 50% of cell viability at 7.5- and 5-fold lower concentrations, respectively. Importantly, none of the investigated compounds caused a reduction in the viability of the nonmalignant human embryonic kidney cells (HEK293) at concentrations up to 50 µM. Next, we investigated the antiproliferative activity of 20d and 19b in HCT-116 cells using clonogenic assay and observed that both compounds effectively inhibited the formation of colonies in a concentration-dependent manner in this cell line (Figure 4c and Figure 4d).
Table 1. In vitro anticancer activity of investigated compounds (IC50±SD (µM)) towards human colon cancer (HCT-116), non-small cell lung adenocarcinoma (A-549), and human breast carcinoma (MCF-7). IC50 value represent a concentration that inhibits 50% of cell growth.
|
Compound
|
Cell lines
|
|
HCT-116
|
A-549
|
MCF-7
|
HEK293
|
|
IC50 [µM]
|
|
9
|
>50
|
>50
|
>50
|
>50
|
|
16
|
>50
|
>50
|
>50
|
>50
|
|
17
|
>50
|
>50
|
>50
|
>50
|
|
18a
|
39.83±2.62
|
27.24±1.53
|
>50
|
>50
|
|
18b
|
>50
|
>50
|
>50
|
>50
|
|
18c
|
18.93±1.26
|
23.83±4.02
|
>50
|
>50
|
|
18d
|
>50
|
>50
|
>50
|
>50
|
|
18e
|
>50
|
45.33±4.20
|
>50
|
>50
|
|
19a
|
26.17±1.69
|
>50
|
>50
|
>50
|
|
19b
|
13.49±0.20
|
15.69±2.56
|
>50
|
>50
|
|
19c
|
12.96±2.68
|
28.44±0.56
|
>50
|
>50
|
|
19d
|
31.64±0.58
|
41.07±0.93
|
>50
|
>50
|
|
19e
|
13.88±1.30
|
>50
|
>50
|
>50
|
|
20a
|
13.11±1.55
|
21.79±0.22
|
>50
|
>50
|
|
20b
|
>50
|
>50
|
>50
|
>50
|
|
20c
|
18.44±2.04
|
23.83±4.02
|
>50
|
>50
|
|
20d
|
12.04±0.57
|
12.55±0.54
|
>50
|
>50
|
|
20e
|
>50
|
>50
|
>50
|
>50
|
|
21
|
>50
|
>50
|
>50
|
>50
|
|
22
|
>50
|
>50
|
>50
|
>50
|
|
23
|
>50
|
>50
|
>50
|
>50
|
|
24
|
>50
|
>50
|
>50
|
>50
|
|
Cisplatin
|
24.89±1.14
|
29.01±0.12
|
33.80±1.56
|
28.45±1.97
|
Based on their antiproliferative activity, 20d and 19b were evaluated for their effect on cell cycle phase distribution and DNA content by flow cytometry. The compounds induced alterations in cell cycle distribution in HCT-116 cells at every time-point of treatment. In comparison to vehicle, treatment with compounds 20d and 19b for 24 to 72 h led to a time-dependent increase (up to <14%) in the number of cells in the G0/G1 phase (Figure 5). Concomitantly, a substantial decrease in the number of cells in the S-phase as well as negligible change in the sub-G1 and G2/M fractions was observed, especially in the case of 20d.
Considering its interesting in vitro antiproliferative activity and the strong inhibitory effect on cell cycle progression, we selected compound 20d for the further analysis of the mechanism of its molecular action.
20d induces generation of ROS in HCT-116 cells
Reactive oxygen species (ROS) affect the cellular redox homeostasis resulting in extensive and irreparable damage and ultimately cell death31. Anticancer compounds with cytotoxic properties often cause disturbances in the redox state in cells. Therefore, we performed a time-dependent kinetic analysis of the intracellular ROS level in colon cancer cell lines after treatment with 20d. As depicted in Figures 6, the compound induced the generation of a high amount of ROS in HCT-116 cells, but not in A-549 cell line, independently of the exposure time (~30% and 5% increase in ROS level compared to vehicle).
We further attempted to determine whether 20d-induced cancer cell death was related to the induction of ROS production in HCT-116 cells. For this purpose, we pretreated cells with N-acetyl-L-cysteine (NAC), which is a commonly used antioxidant and ROS scavenger. As shown in Figure 6b and Figure 6c, NAC diminished the cytotoxic effect of 20d in HCT-116. This indicated that ROS-dependent cellular redox imbalance is the main mechanism of 20d-mediated cell death in this cell line. HCT-116 cell line has high-frequency microsatellite instability, which is associated with deficient DNA mismatch repair (MMR)32. The MMR system corrects DNA-mismatched and insertion–deletion loop bases generated during DNA replication33. One of the key proteins involved in the MMR pathway is MLH1, which is mutated or epigenetically silenced in many MMR-deficient (MMR–) cells, such as HCT-11634. To further determine the selectivity for MMR deficiency, we analyzed the cell viability of human MutL homologue 1 (hMLH1)-deficient (MMR–) and hMLH1-proficient (MMR+) RKO human colon carcinoma cell lines. The latter was established by transfecting hMLH1 cDNA into MMR− RKO cell line35. In MMR– RKO cells, the hMLH1 gene promoter was silenced transcriptionally by hypermethylation36. The viability assay revealed that MMR− RKO cells were 3.95-fold more sensitive to 20d (p<0.001) compared to the MMR+ RKO cells (Figure S55 in supplementary information), and the respective IC50 values (mean±SD) were 1.21±0.13 and 4.78±0.38 μM.
20d oxidative properties are related to MMR deficiency
Then, we confirmed whether the induction of cellular stress by 20d is related to MMR deficiency by exposing MMR-deficient and MMR-proficient cells to the compound for different times and measuring the ROS levels. Our results indicated a greater increase in the level of ROS after 1 h of exposure in 20d-treated MMR-deficient cells than in MMR-proficient cells (54.71±2.70% and 22.27±1.7%, respectively), in comparison to control (Figure 7). This finding suggests that 20d could modulate oxidative stress in human MLH1-defective colon cancer cells through synthetic lethality.
20d treatment induces autophagy cell death in HCT-116 cells through the PI3K/AKT/mTOR signaling pathway
Due to its ability to mediate the redox signaling pathways, ROS interplays between two major types of programmed cell death: autophagy and apoptosis37. One of the early events in the process of apoptosis is the translocation of phosphatidylserine to the exposed membrane surface, which can be detected using Annexin V, a protein with a high affinity to this phospholipid38. Flow cytometry analyses with double-staining using Annexin V-fluorescein isothiocyanate (FITC) conjugate and prodidium ioidide (PI) revealed that 20d induced apoptosis- and necrosis-independent cell death in HCT-116 cells (Figure S55 in supplementary information).
Then, to elucidate whether compound 20d can act as an autophagy modulator, we stained cells with acridine orange (AO), a lysotropic agent39. AO is a weak base that can permeate the cell membrane in an uncharged state emitting green fluorescence. After protonation, AO forms aggregates in acidic vesicular organelles (AVOs), emitting red fluorescence33. As shown in Figure 8a and Figure 8b, treatment with 20d significantly enhanced the accumulation of AVOs in HCT-116 cells, resulting in a 1.9-fold (p<0.01) increase in red fluorescence in comparison to vehicle. One of the hallmarks of autophagy is the conjugation of LC3-I with phosphatidylethanolamine, followed by its conversion to autophagosome-associated form LC3-II40. Taking this into account, we evaluated the expression of LC3I/II in colon cancer cells after exposure to 20d. Furthermore, we studied the expression of LAMP-1, a well-known lysosomal marker41. Immunoblot assays revealed that the levels of LC3-II were higher in 20d-treated HCT-116 cells compared to control groups (Figure 8c and Figure 8f). This observation was also confirmed by increased punctate cytosolic LC3 fluorescence intensity when HCT-116 cells were assessed by confocal microscopy (Figure 8e and Figure 8g). In addition, a time-dependent increase in the expression of LAMP-1 protein was noted after 20d treatment. It has been shown that multiple signaling pathways are involved in the modulation of autophagy, including the PI3K/AKT/mTOR pathway, which is especially involved in the stage of autophagosome elongation42,43. In this study, we investigated whether 20d-induced autophagy in HCT-116 cells is related to PI3K/AKT/mTOR signaling. The results showed that 20d significantly reduced the levels of phospho-AKT (Ser473) and phospho-mTOR (Ser2448) proteins, in a time-dependent manner (Figure 8d and Figure 8f). This proves that 20d induced autophagy cell death in HCT-116 cells by interfering with the PI3K/AKT/mTOR signaling pathway.
20d modulates expression of the cell cycle-related protein
To understand the mechanisms underlying 20d-induced cellular death in detail, we estimated the expression levels of cyclin D1, p21, p27, and p53 in HCT-116 cells by Western blot assay for the indicated time (Figure 9). p21 is a negative regulator of the cell cycle, which mediates many cellular processes primarily by inhibiting the activity of cyclin-dependent kinase (CDK) 2 and CDK1 (also known as CDC2), leading to growth arrest44. p27, another tumor suppressor, halts the cell cycle via CDK complexes, while also playing a role in survival, differentiation, and migration45.
As shown in Figure 9, the expression of p53 and cell cycle regulatory proteins p21 and p27 were markedly higher in 20d-treated cells (p<0.001) than in the control group. Oxidative stress leads to the stabilization of p53, which is then activated to promote cell cycle arrest and induce autophagy by transcriptionally activating target genes, such as DNA damage-regulated autophagy modulator 1 (DRAM1)46 and Sestrin 1/2 (SESN1 and SESN2)47. While upregulation of proteins p21 and p27 causes halting of G1/S-phase transition, transcription factor p53 upregulates many genes in response to double-strand breaks (DSBs), including proto-oncogene MYC, which codes for the transcription factor c-Myc48. Therefore, we analyzed the expression of c-myc after treatment with 20d and found that it was downregulated in the treated cells as compared to vehicle. This suggests that p53 negatively regulates c-myc. Suppression of MYC may cause tumor cells to lose their neoplastic properties, because of diverting cellular resources toward stress response pathways, permitting cells to recognize DNA damage, leading to enforces their regression and differentiation48.
20d induces DNA damage in HCT-116 cells
ROS formation can induce DNA oxidation followed by mutagenic alterations in DNA bases or double-helix breaks, resulting in cell cycle arrest and subsequently cell death49. Therefore, we further investigated the effect of 20d on DNA damage response. HCT-116 cells were incubated with 20d for 24–72 h, and then the expression of p-γ-H2AX, a known DNA damage marker, was determined by immunoblot and immunofluorescence. As shown in Figure 9 and Figure 10, even after 24 h, the expression of p-γ-H2AX was found to be significantly increased. It was also noted that 20d led to this increase in p-γ-H2AX in a time-dependent manner.
β-Catenin is a member of the Armadillo repeat protein family and an important component of the cell–cell adhesion machinery. It is also involved in the Wnt growth factor signaling pathway50,51. As shown in Figure 9, 20d caused the cleavage of β-catenin, even after 24 h of exposure. Proteolysis of β-catenin has often been observed during apoptotic cell death. However, some reports indicate that such proteolysis may also occur due to the disorganization of endothelial adhesion junctions52,53,54. To investigate whether β-catenin regulates the expression of cyclin D1 in colon carcinoma cells, we determined its expression after 20d treatment55. We noted a slight inhibition of cyclin D1 expression in 20d-treated cells compared with the DMSO-treated cells after 72 h of treatment, which might be associated with its induction of growth arrest in colon cancer cells. Additionally, we analyzed the changes in the expression of vimentin, another protein known to be involved in cell–cell junctions and thus contribute to epithelial–mesenchymal transition56. As shown in Figure 9, the expression of vimentin was significantly altered in 20d-treated cells.
Suppression of HCT-116 cells migration following 20d treatment
A decrease in cell–cell adhesion correlates with tumor invasion and metastasis57. Migration leads to spreading and metastasis of cancer cells and is thus associated with poor prognosis of many types of cancer58. Moreover, studies indicate that metastasis is the major cause of death in patients with colorectal cancer (CRC)59. To investigate the effect of 20d on migration, HCT-116 cells were cultured and investigated by wound healing assay. The percentage of migration was monitored over time by capturing a series of images after every 6 h up to 36 h of treatment. As shown in Figure 10c and Figure 10d treatment with 20d led to a significant reduction (p<0.0001) in migration in HCT-116 cells, compared to DMSO-treated vehicle. This antimigratory effect was more evident at 30 and 36 h of treatment, during which a 30–50% inhibition of migration was observed in HCT-116 cells treated with 10 and 20 µM 20d. Importantly, we did not observe any detached cells after treatment, which indicates that exposure to 20d did not cause cell death at any tested time-point. In addition, as shown in Figure S65 in the supplementary information, the compound 2md shows a slight cytotoxic effect after 24h and 48h exposure at concentrations of 10 and 20 µM, thus observed antimigratory properties of investigated compound is not affected by cell death.
{kind=link}
{kind=link}