Overview of the sequencing data
We constructed cDNA libraries of miRNAs, mRNAs, and lncRNAs using the RNA from the ovaries and testes. After filtering out low-quality transcripts, 5′ and 3′ adapters, and reads < 18 nt, a total of 113.5 M of clean reads was produced by Illumina technology for miRNAs. The 21 and 22 nt length transcripts were the most abundant (Fig. 1a), and 60.4% of high-quality reads were mapped to the turtle genome (Pelsin-1.0, NCBI). We obtained 153.25 Gb of clean reads for mRNA and lncRNA sequencing. The length distributions of lncRNAs and mRNAs are shown in Fig. 1b and 1c. After mapping the genome, approximately 84.41% ~ 87.72% of the reads were mapped to intergenic regions in the P. sinensis reference genome (Fig. 1d, Additional 1).
Identification of the differential expression of mRNAs, miRNAs, and lncRNAs
According to the miRNA expression profiles, we detected 10 446 novel miRNAs. A total of 633 miRNAs were significantly differentially expressed between the ovaries and testes (P < 0.05), including 138 up-regulated miRNAs and 495 down-regulated miRNAs (Fig. 2a, 2d, Additional file 2). These DEmiRNAs belonged to 438 families (Additional file 3). Among these DEmiRNAs, we identified a set of miRNAs that were reported to regulate animal reproduction, including miR-133, miR-138, miR-145, miR-143, and miR-378.
We detected 20 414 mRNAs, and 11 319 mRNAs were differentially expressed based on sex, including 5 206 up-regulated mRNAs and 6 113 down-regulated mRNAs (Fig. 2b, 2e, Additional file 4). A total of 28 500 lncRNAs with 10 495 DElncRNAs were detected, including 1 716 up-regulated lncRNAs and 8 779 down-regulated lncRNAs between ovaries and testes (Fig. 2c, 2f, Additional file 5). Among the differentially expressed lncRNAs and miRNAs, 3 miRNAs and 917 lncRNAs exhibited testis-specific expression, and 186 miRNAs and 4 491 lncRNAs showed ovary-specific expression. Prediction of the potential targets of lncRNAs in cis and trans was performed to investigate the function of lncRNAs (Additional file 5). After searching for protein-coding genes 100 kb upstream and downstream, 3 904 DElncRNAs were found to correspond to the regulation of protein-coding genes in cis. The target genes included Foxl2, Cyp19a1, Gper, Esr, Dazl, and Sox30, which suggests that the reproductive process might be regulated by the action of these lncRNAs on protein-coding genes. Conversely, we identified 2 160 lncRNAs showing trans action by LncTar, including a set of genes that might regulate reproduction.
Functional analysis of DEmiRNAs and DElncRNAs
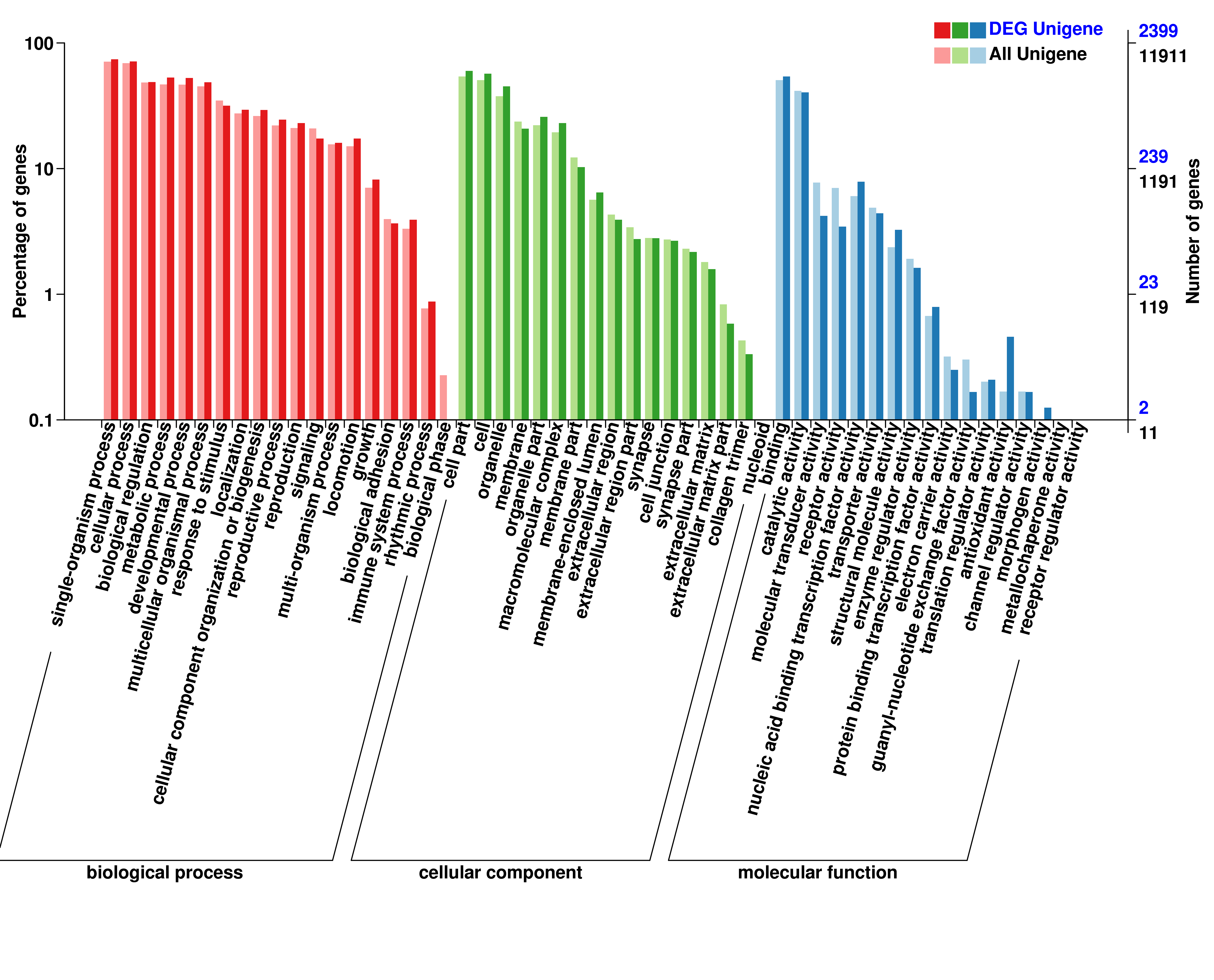
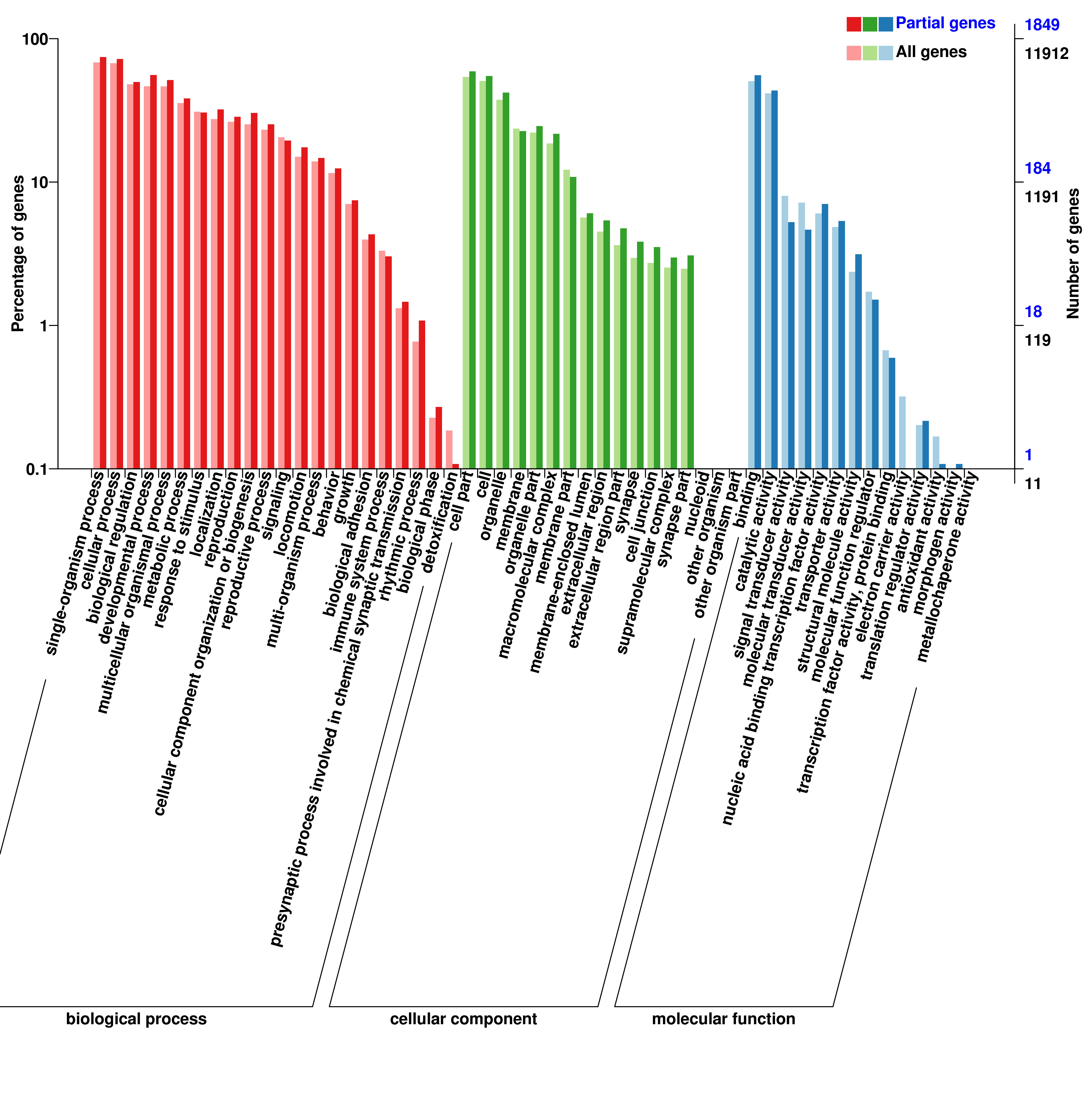
To annotate the molecular functions of the differentially expressed miRNAs, both RNA hybrid and MiRanda software were used to improve the prediction of miRNA targets, resulting in 8 088 target genes including 2 814 differentially expressed genes that were potentially regulated by 633 DEmiRNAs. GO categories of miRNAs and lncRNAs were assigned to all target genes based on the following three ontologies: cellular component, molecular function, and biological process (Additional file 6, 7, 8). Functions of target genes in the cellular component category mainly focused on cell part, cell, and membrane. Based on molecular function, the most abundant target genes were focused on binding, followed by catalytic activity. Regarding biological process, the most abundant target genes were focused on single organism process, followed by cellular process, and biological regulation.
KEGG pathway enrichment analysis revealed that the DEmiRNAs were involved in 186 signalling pathways (Additional file 9). The identified metabolic networks were related to neuroactive ligand-receptor interaction and regulation of the actin cytoskeleton. The most abundant target genes of DEmiRNAs focused on glyoxylate and dicarboxylate metabolism. We detected at least 13 pathways involved in reproductive biology, including oocyte meiosis, TGF-β signalling, ovarian steroidogenesis, GnRH signalling, Wnt signalling, cAMP signalling, steroid biosynthesis, steroid hormone biosynthesis, MAPK signalling, p53 signalling, RNA polymerase, metabolism of xenobiotics by cytochrome P450, and mTOR signalling.
KEGG pathway enrichment analysis showed that the DElncRNAs were involved in 225 signalling pathways in a trans-regulatory manner and 221 signalling pathways in a cis-regulatory manner (Additional 10, 11). The KEGG pathway enrichment analysis revealed that the DElncRNAs were involved in oocyte meiosis, steroid hormone biosynthesis, Wnt signalling pathway, GnRH signalling pathway, p53 signalling pathway, apoptosis, MAPK signalling pathway, AMPK signalling pathway, TGFβ signalling pathway, cAMP signalling pathway, RIG-I-like receptor signalling pathway, mTOR signalling pathway, and insulin signalling pathway.
Validation of differentially expressed miRNAs and lncRNAs
To validate the sequencing data of miRNAs and lncRNAs, ten DEmiRNAs and ten DElncRNAs were randomly selected to test their relative expression in ovaries and testes. The expression of eight miRNAs and seven lncRNAs in ovaries and testes was consistent with the results of RNA sequencing. Among the miRNAs, novel-miR-1361, novel-miR-2322, novel-miR-6721, novel-miR-10042, novel-miR-10231, novel-miR-10322, and novel-miR-10468 were downregulated in testis, while novel-miR-1236 was upregulated in testes (Fig. 3a). Among the lncRNAs, MSTRG.435295.1, MSTRG.88998.1, MSTRG.127189.1, and MSTRG.100955.1 were upregulated in testes, while MSTRG.129036.2, MSTRG.281180.2, and MSTRG.561412.1 were downregulated in testis (Fig. 3b). The expression patterns of these miRNAs and lncRNAs among different groups were well-matched with the RNA-Seq data, which could guarantee the accuracy of subsequent functional analysis.
Construction of compete endogenous (ceRNA) networks
To construct the ceRNA networks, we screened miRNAs that included miRNA response elements, which could bind with both lncRNAs and mRNAs. We constructed a series of ceRNA networks of mRNAs, miRNAs, and lncRNAs related to the DE genes by integrating the expression profiles and regulatory relationships among the mRNAs, lncRNAs, and miRNAs from the high-throughput sequencing data (Additional file 12). These networks included 102 DEmiRNAs, 635 DEmRNAs, and 1 621 DElncRNAs. The DEmiRNAs included novel-miR-227, novel-miR-9914, novel-miR-6375, novel-miR-1222, novel-miR-6721, novel-miR-2026, novel-miR-6671, novel-miR-642, novel-miR-6319, and novel-miR-42, etc. These ceRNA networks included a set of mRNAs regulating reproduction (Fig. 4a, 4b, Additional file 12). For instance, Dazl mRNA and MSTRG.71049.8 shared a common binding site of the miRNA novel-miR-1222. We also identified Wt1, CREB3l2, Gata4, Wnt2, Nr5a1, Hsd17, Igf2r, H2afz, Lin52, Trim71, Zar1, and Jazf1 in the ceRNA network. These miRNAs and mRNAs participate in regulating the reproductive process, including meiosis and spermatogenesis.
{kind=link}
{kind=link}
{kind=link}