Identification of cotton hybrids exhibiting different level of heterosis
Our previous study of 11 inbred and 30 hybrids in six environments showed that cotton hybrids had better and stable performance compared with inbred parents in yield traits [40]. Here, analysis of heterosis revealed the majority of hybrids had positive mid parent heterosis (MPH) and better parent heterosis (BPH) for boll weight, seed cotton yield (SCY), lint yield and lint percentage (Table S1). Later on, three hybrids with different level of heterosis were identified and defined as high (H), medium (M) and low (L). Notably, H-hybrid (SJ48×Z98) produced 19.9% MPH and 14.1% BPH in SCY (Fig. 1). M-hybrid (SJ48×851) had MPH of 13.3% and BPH of 2% for SCY, whereas L-hybrid (SJ48×DT) showed the only MPH of 8.8% for SCY. These three hybrids had common maternal inbred parent but paternal inbred parents all differed. H-hybrid is cross of maternal line SJ48 (A) with paternal parent Z98 (B) and M-hybrid with paternal parent 851 (C), whereas L-hybrid is cross of same maternal line with paternal parent DT (D). The differences in the level of yield heterosis suggest that these three hybrids together with their inbred parents are suitable for studying comparative transcriptome analysis and regulatory mechanism of yield heterosis in cotton.
Transcriptome profiles of 63 RNA libraries of cotton hybrids and their inbred parents at squaring stage
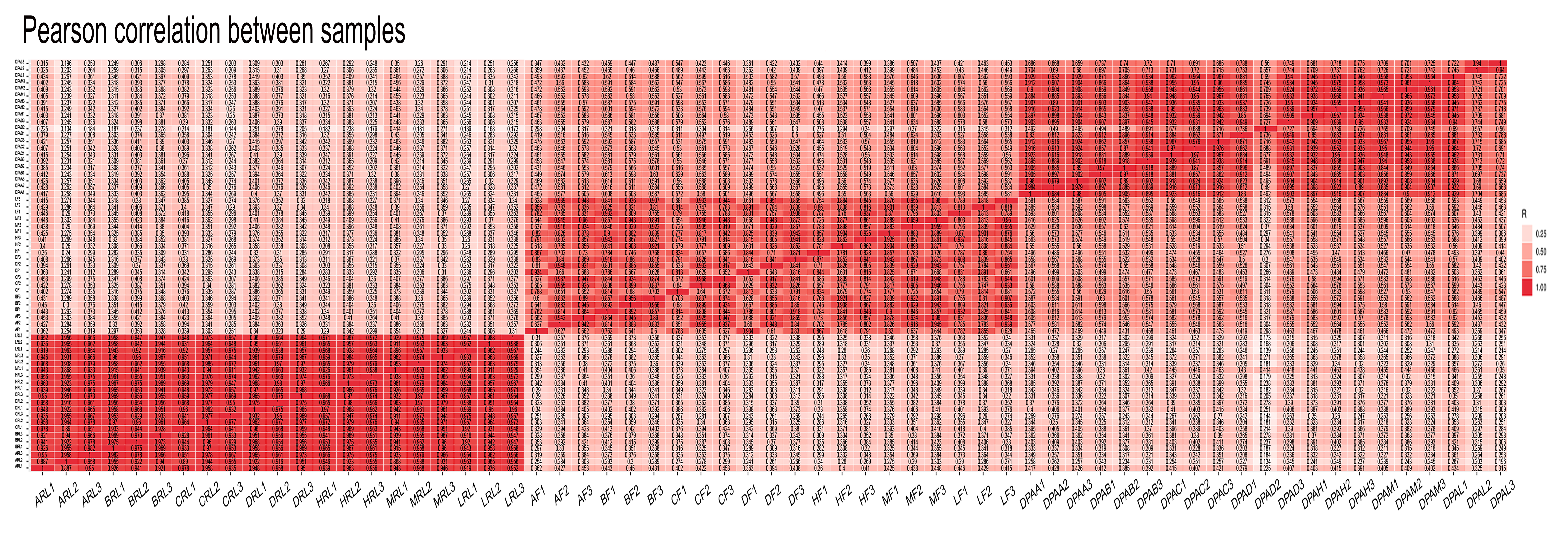
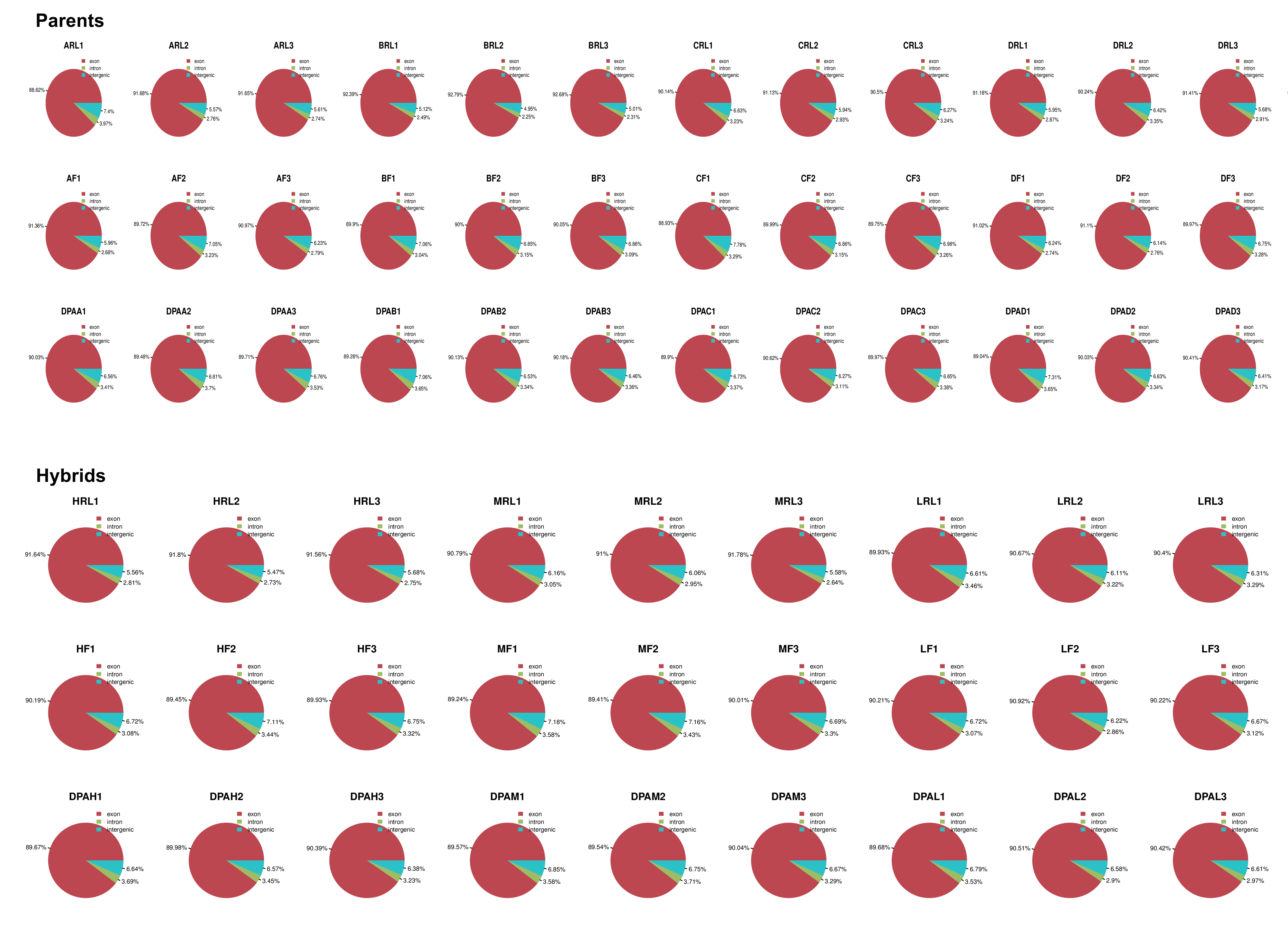
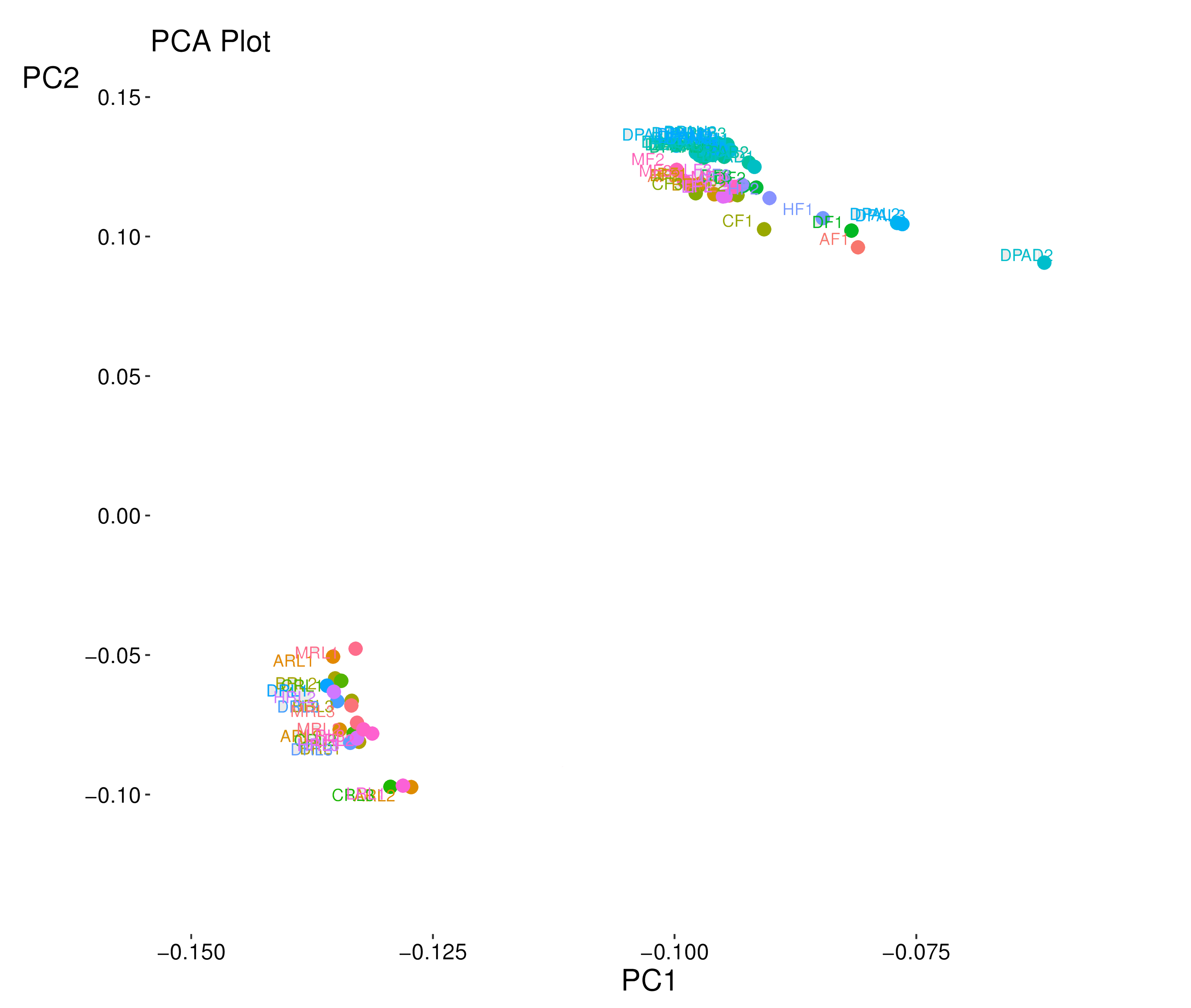
To understand global transcriptome profiles of yield heterosis in cotton, three contrasting hybrids and their four inbred parents were used to perform RNA sequencing at the initiation of squaring stage. Leaf (Hereafter RL), flower buds (F), and one day post-anthesis (1DPA) ovule tissues with three biological replications were used for each genotype. In total, 63 libraries (7×3×3) were constructed for deep Illumina pair-end RNA sequencing. A total of ~43-50 million reads were generated per library (Table 1). The average percentage of the valid read was ≥98.5 %. The value of Q30% was above 95% in this sequencing. Approximately, 94.9% of clean reads were mapped to the reference G. hirsutum genome [36]. Almost 90.5% reads were mapped to exon region in each sample and genotype. Wherein, intron and intergenic region mapping were ≤5% (Fig. S1). The brief detail of total mapped, unique mapped, multi mapped, non-splice, and splice reads for each library can be seen in Table 1. The principal component analysis revealed two main clusters of all samples based on the two-first PCs (Fig. S2). RL replicates of most genotypes were found in one clustered with less variation, whereas tissues of F and 1DPA had distinct clusters. Pearson correlation test for all replicate and genotypes exposed that RL tissue had a strong correlation among each other (≥75) and weak with F and 1DPA (≤ 50) (Fig. S3). Correlation between F and IDPA samples was relatively strong (~55-60).
Global transcriptome changes for cotton hybrids and their inbred parents
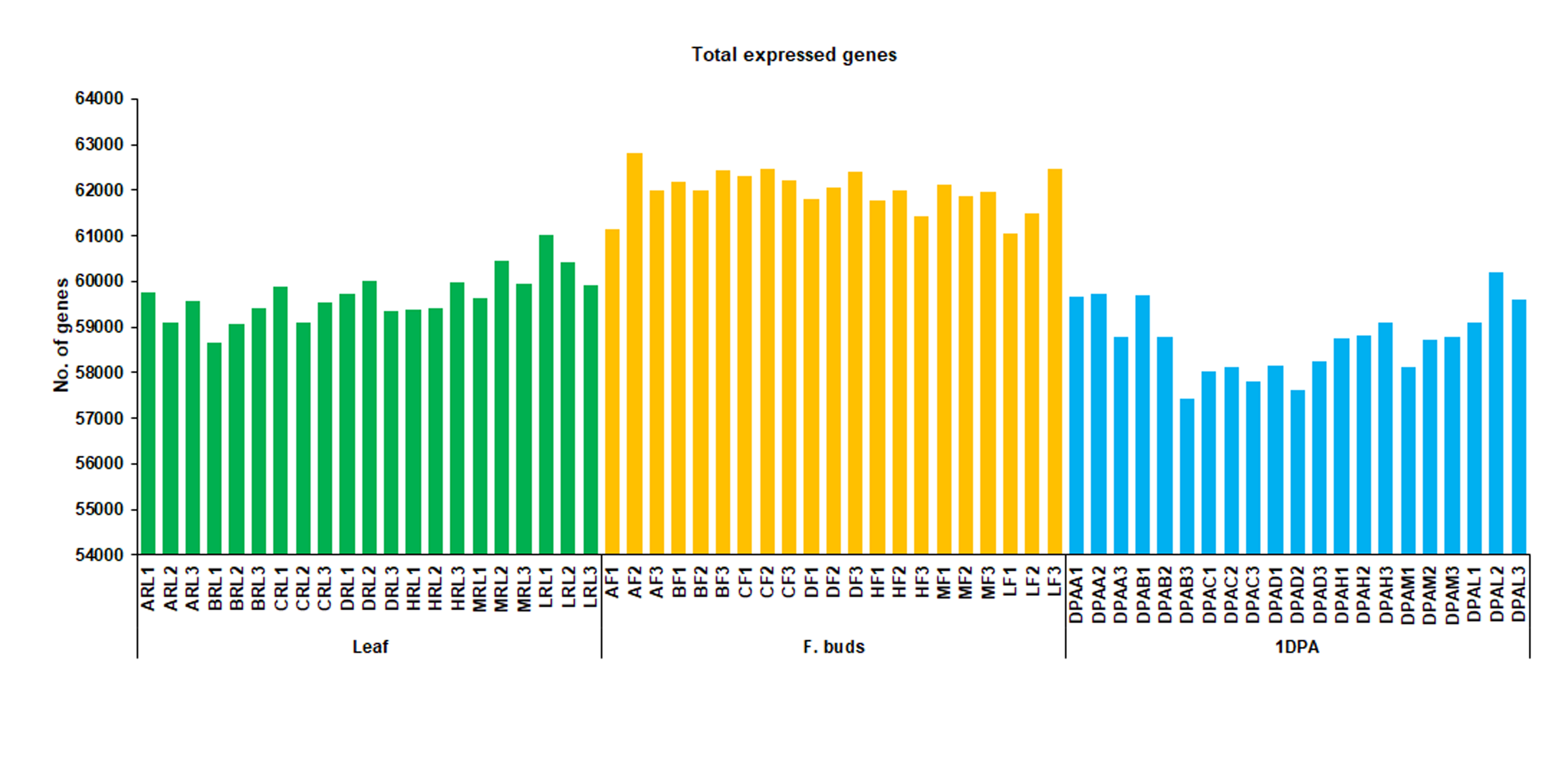
The number of total expressed genes provides an overview of the transcriptomic landscape for all datasets. A total of ~60000 genes were expressed in each dataset (Fig. S4). Total expressed genes were much higher in flower buds compared to leaf and 1PDA ovule, while 1DPA ovule showed fewer total expressed genes compared with the other two tissues. Gene expression levels calculated as FPKM were used to analyze the dynamic changes of transcriptomes between hybrids and their respective parents. The expression levels significantly different at p ≤ 0.05 and with log2 (fold change) >1 or <-1 designed as differentially expressed genes (DEGs). Here, the comparison of transcriptomes was performed for each hybrid parent triad and tissue. Total number of DEGs (Up + down) and their distribution among H-hybrid parent triad is shown in Fig. 2. Comparative analysis among H and its maternal parents (A) showed a total of 1121 DEGs in leaf (RL), 2073 in flower buds (F), and 1202 in 1DPA ovule, while compared with paternal parent (B), 1129, 1514, and 1805 DEGs were respectively identified in RL, F, and 1DPA (Fig. 2a). Combination of both parents A and B displayed higher number of DEGs (~2100 in all tissues) relative to H-hybrid. Distribution of DEGs found major portion were common in hybrid parent triad, whereas less were unique in each tissue (Fig. 2b, c, d). For example, 367 DEGs in RL, 775 in F, and 473 in 1DPA were unique in the combination of A with H. Unique DEGs in comparison of B and H were 358 in RL 577 in F, and 624 in 1DPA.
The results of DEGs in RL, F, and 1DPA transcriptomes of M-hybrid parent triad revealed that comparison of M with its maternal parent (A) had 1049, 1116, and 1207 DEGs respectively (Fig. 3a). Compared with paternal parent (C), this displayed 1518 DEGs in RL, 1471 in F, and 1196 in 1DPA. Combination of both parents A and C revealed a total of 1258, 1395, and 1576 differential gene expressions in RL, F, and 1DPA respectively. Distribution of unique and common DEGs identified more commonality in each tissue (Fig. 3b, c, d). Fewer DEGs were unique in A versus M such as 365 in RL, 405 in F, and 466 in 1DPA, while these were 626 in RL, 511 in F, and 376 in 1DPA in combination of C versus M. In context of commonality, both A and C compared with M had 362 overlap DEGs in RL, 375 in F, and 305 in 1DPA. Comparative analysis of DEGs for L-hybrid parent triad is summarized in Fig. 4. The result revealed that L versus maternal parent (A) had 1112 total DEGs in RL, 935 in F, and 3528 in 1DPA (Fig. 4a). The comparison of L and paternal parent (D) showed 1524, 790, and 4118 total number of DEGs in RL, F, 1DPA, respectively. Comparisons among parents A and D as compared to L displayed higher number of DEGs in RL (1626) and F (1415) and much less in 1DPA (2027). Distribution of DEGs revealed that majority of genes were overlapped but less were unique in each tissue (Fig. 4b, c, d). A versus L had 346, 299, and 1101 unique DEGs in RL, F, and 1DPA, respectively. Unique DEGs in D versus L were 649 in RL, 256 in F, and 1357 in 1DPA. In the context of commonality, comparison of L with both A and D showed 304, 181, and 1963 shared DEGs in RL, F, and 1DPA, respectively. At squaring stage, comparative transcriptome analysis in each hybrid parent triad determined that the percentage of genetic differential expressions in hybrids was similar to those of between parents.
Analysis of expression level dominancy for hybrids
Expression level dominance is a phenomenon in which expression of hybrid DEGs is statistically similar to that of one parent, maybe additive or transgressive. To identify the expression magnitude and directionality in interspecific F1 hybrid cotton, DEGs were divided into 12 possible groups as described in material and methods following the conventions of [41] (Fig. 5a). Results showed that the magnitude of expression of most genes in hybrids was similar to that of one parent (Fig. 5). For instance, most of the genes in leaf and 1DPA ovule of H-hybrid displayed higher-maternal dominance expression and lower-maternal dominance expression respectively (Fig. 5b; Fig. S5a, c). Lower-paternal dominance expression had highest proportion in flower buds (Fig. 5b; Fig. S5b). Non-additive expressed genes in leaf and 1DPA ovule of M-hybrid mostly displayed higher-maternal dominance expression (Fig. 5c; Fig. S6a, c), wherein lower-maternal dominance expression was highest in flower buds (Fig. 5c; Fig. S6b). In the case of L-hybrid, most of leaf genes showed higher-maternal dominance expression (Fig. 5d; Fig. S7a), while lower-paternal dominance expression had highest portion in flower buds (Fig. 5d; Fig. S7b). In parent like expression groups, most of the genes in IDPA ovule displayed higher maternal dominance expression in this hybrid (Fig. 5d; Fig. S7c). Accordance with analysis of expression level dominance, most DEGs of hybrids displayed parent expression level dominancy (P-ELD) at squaring stage in different tissues that probably play role for yield heterosis of cotton.
Functional annotation and pathway enrichment analysis of DEGs with P-ELD
To identify the function of DEGs with P-ELD, a total of four gene sets (3-6 groups) were pooled to perform GO and KEGG enrichment analysis for every hybrid and tissue (Table S2, S3). GO enrichment analysis showed that majority of genes in leaf of H-hybrid had functional annotation related to biological process, integral component of membrane, ATP and protein binding (Fig. 6a). Most abundant functional gene groups in flower buds were plasma membrane, integral component of membrane and ATP binding, wherein nucleus, plasma membrane, and protein binding genes had higher significance in 1DPA ovule (Fig. 6a). Genes associated with chloroplast, extracellular region, and ATP binding had highest portion in leaf of M-hybrid (Fig. 6b). In flower buds, most of the genes were linked with biological process, molecular function and integral component of membrane. Most abundant functional gene groups in 1DPA ovule were biological process, chloroplast, and mitochondrion (Fig. 6b). Most enriched GO terms in leaf of L-hybrid were ATP binding, serine/threonine kinase activity (Fig. 6c). Functional annotation related to biological process, integral component of membrane, and chloroplast were abundant in flower bud, whereas gene involved in plasma membrane, regulation of transcription, and mitochondrion had highest portion in 1DPA ovule of L-hybrid (Fig. 6c).
KEGG enrichment analysis of P-ELD genes exposed that in leaf of H-hybrid starch and sucrose metabolism, endocytosis, and tryptophan metabolism had more significance (Fig. 7a). Majority of genes in flower buds were enriched in starch and sucrose metabolism, phagosome, and pentose and glucuronate interconversions. Whereas plant-pathogen interaction and plant hormone signal transduction were more representative pathways in 1DPA ovule (Fig. 7a). Leaf of M-hybrid showed more enrichment in metabolic pathways e.g. starch and sucrose, ascorbate and aldarate, and tryptophan (Fig. 7b). In flower buds, starch and sucrose metabolism, plant hormone signal transduction had more gene enrichment (Fig. 7b). Starch and sucrose metabolism and phosphatidylinositol signaling system was enriched and significant in 1DPA of this hybrid (Fig. 7b). Pathway categorization showed that L-hybrid had starch and sucrose metabolism and ribosome significance in leaf (Fig. 7c), whereas starch and sucrose metabolism, plant hormone signal transduction, and ribosome were more representative pathways in flower buds (Fig. 7c). In 1DPA ovule, plant hormone signal transduction, plant-pathogen interaction, and starch and sucrose metabolism had more gene enrichment in this hybrid (Fig. 7c).
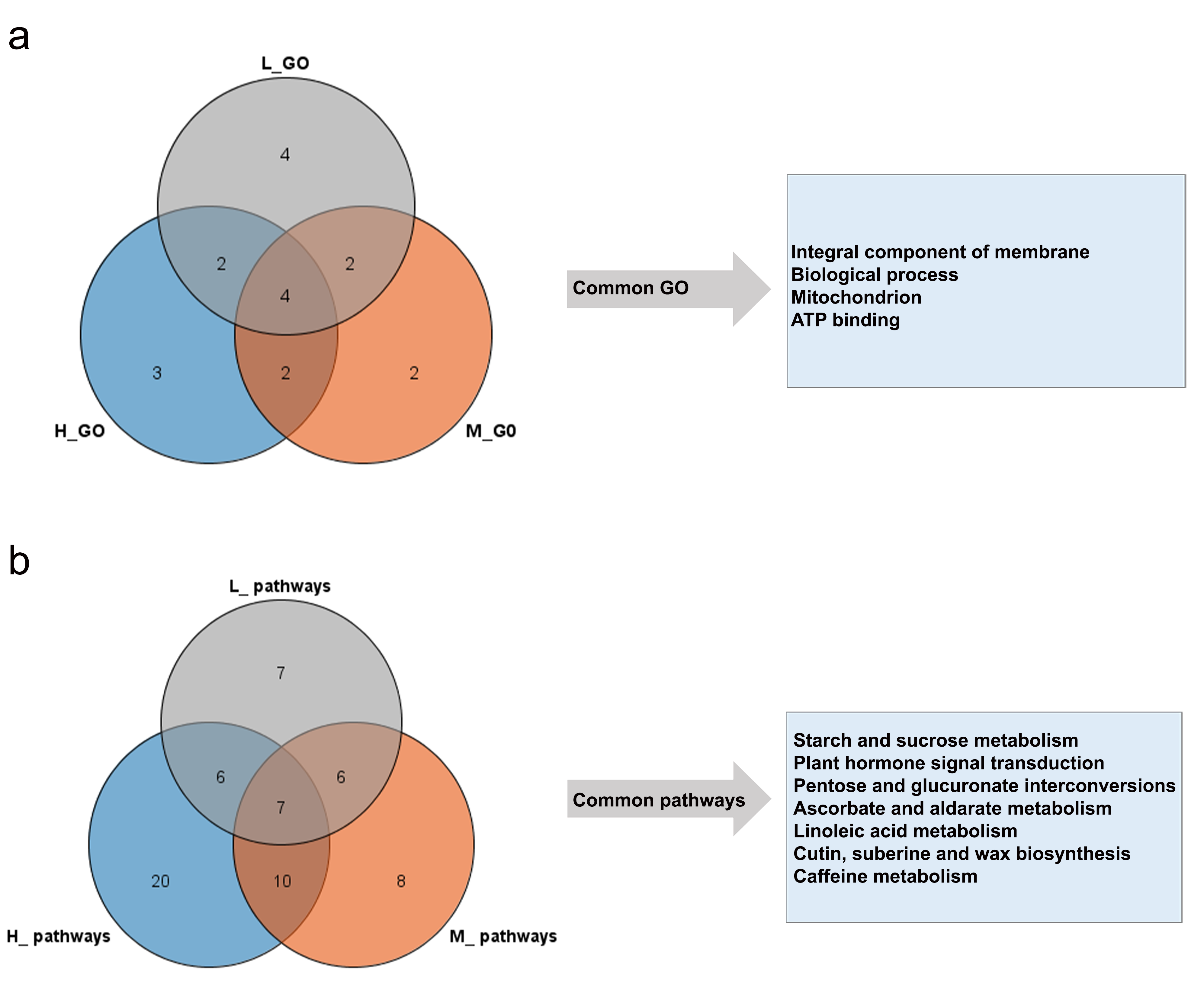
The common function of P-ELD genes in hybrids was integral component of membrane, biological process, mitochondrion, and ATP binding (Fig. S8a). Genes involved in common pathways such as starch and sucrose metabolism, plant hormone signal transduction, pentose and glucuronate interconversions, linoleic acid metabolism, and cutin, suberine and wax biosynthesis seems had a key role in hybrids growth during reproductive stage (Fig. S8b). Theoretically, genes from these pathways contribute to the yield of heterosis in hybrid cotton.
Mapping of key pathways DEGs on known seed cotton yield QTLs
QTL provide associations between genomics and phenomics for complex traits. Using QTLs can be an effective approach to understand the genetic complexities of yield heterosis. Here, we investigated the relationship between key pathway DEGs, seed cotton yield QTLs and heterosis. DEGs of high, medium, and low hybrid compared to their parents, and involved in key pathways such as starch and sucrose metabolism, plant hormone signal transduction, pentose and glucuronate interconversions, linoleic acid metabolism, and cutin, suberine and wax biosynthesis were used to map on already known 57 seed cotton yield (SCY)_QTLs (Table S4). Mapping results showed that 74 hybrid DEGs from these pathways were mapped on 43 QTLs (Fig. 8). Interestingly, most of the genes were mapped on QTLs regions that were reported more than once (Fig. 8). Out of these DEGs, 6 genes had differential expression in all hybrids compared to their parents, whereas 13 were common between any of two hybrids. 13 DEGs were specific to medium hybrid. However, 17 and 25 DEGs were specific to high and low hybrid respectively (Fig. S9)
Co‑expression network analysis of high and low hybrids
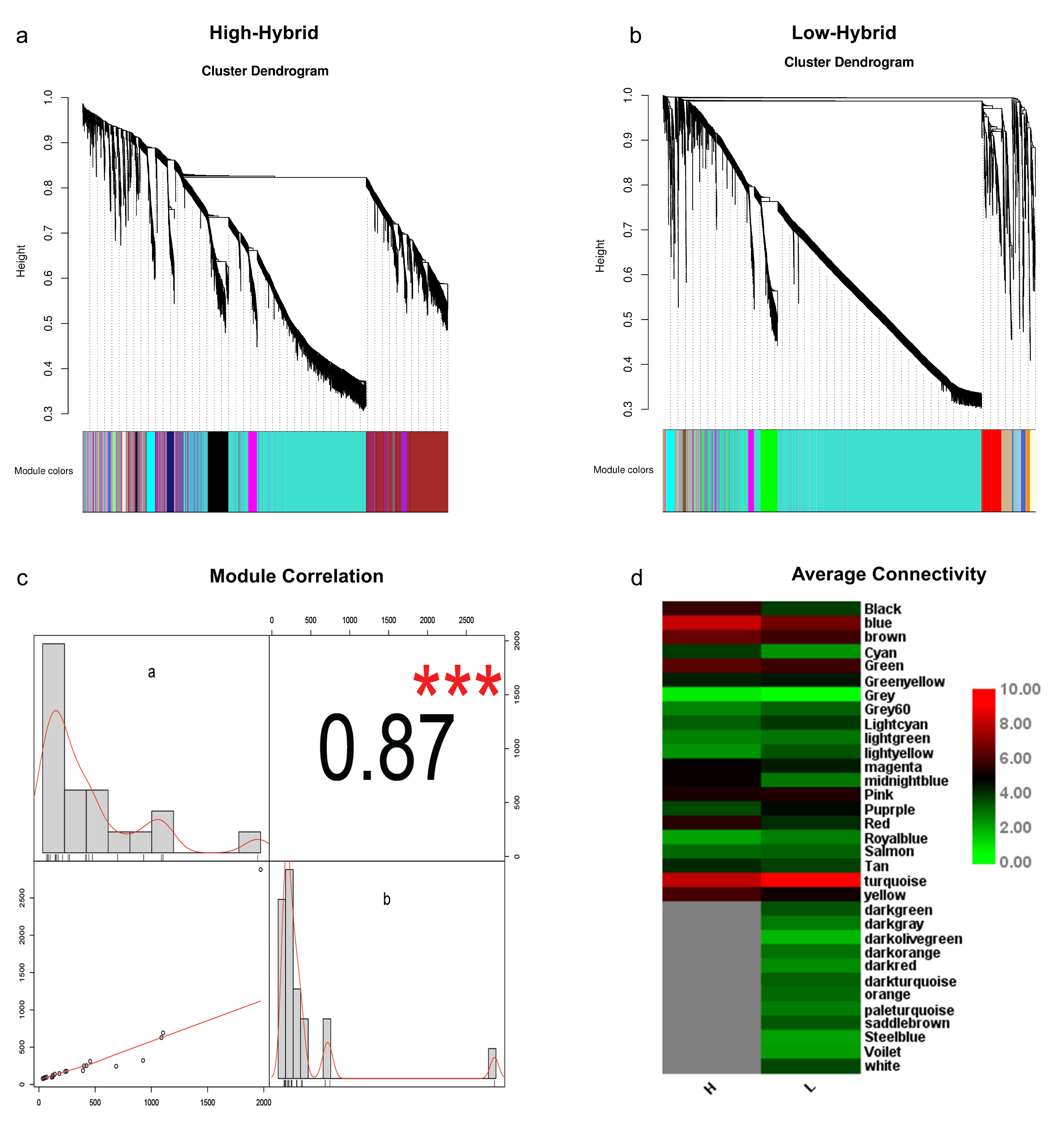
WGCNA is an important tool for inferring the potential relationships between co-expressed genes in different samples or conditions. Construction of co-expression network helps to understand the functional linkage between gene groups and provides novel insight into the system-level understanding of a biological process. Here, co-expression network analysis was performed in high and low hybrids by using DEGs (Pooled from leaf, flower buds and 1DPA) identified in our RNA-seq data analysis. Highly connected genes were clustered into distinct modules. Cluster dendrogram displayed that interconnectivity and size of each model were quite different in high and low hybrids (Fig. S10a, b). Interestingly, 8862 genes distributed in 21 modules with the size ranging from 34 to 1972 genes was identified in high hybrid, wherein 7863 genes scattered in 33 modules with the module size ranging from 42 to 2880 was determined in low hybrid (Table S5). The correlation analysis between high and low hybrids modules was very strong (Fig. S10c). The average connectivity of genes in most of the modules was higher in high hybrids compared with low hybrid (Fig. S10d). Although turquoise and blue module had the highest connected genes in both hybrids, but correlation coefficients among different genes were much higher in blue module.
Genes regulatory co-expression network for seed cotton yield heterosis
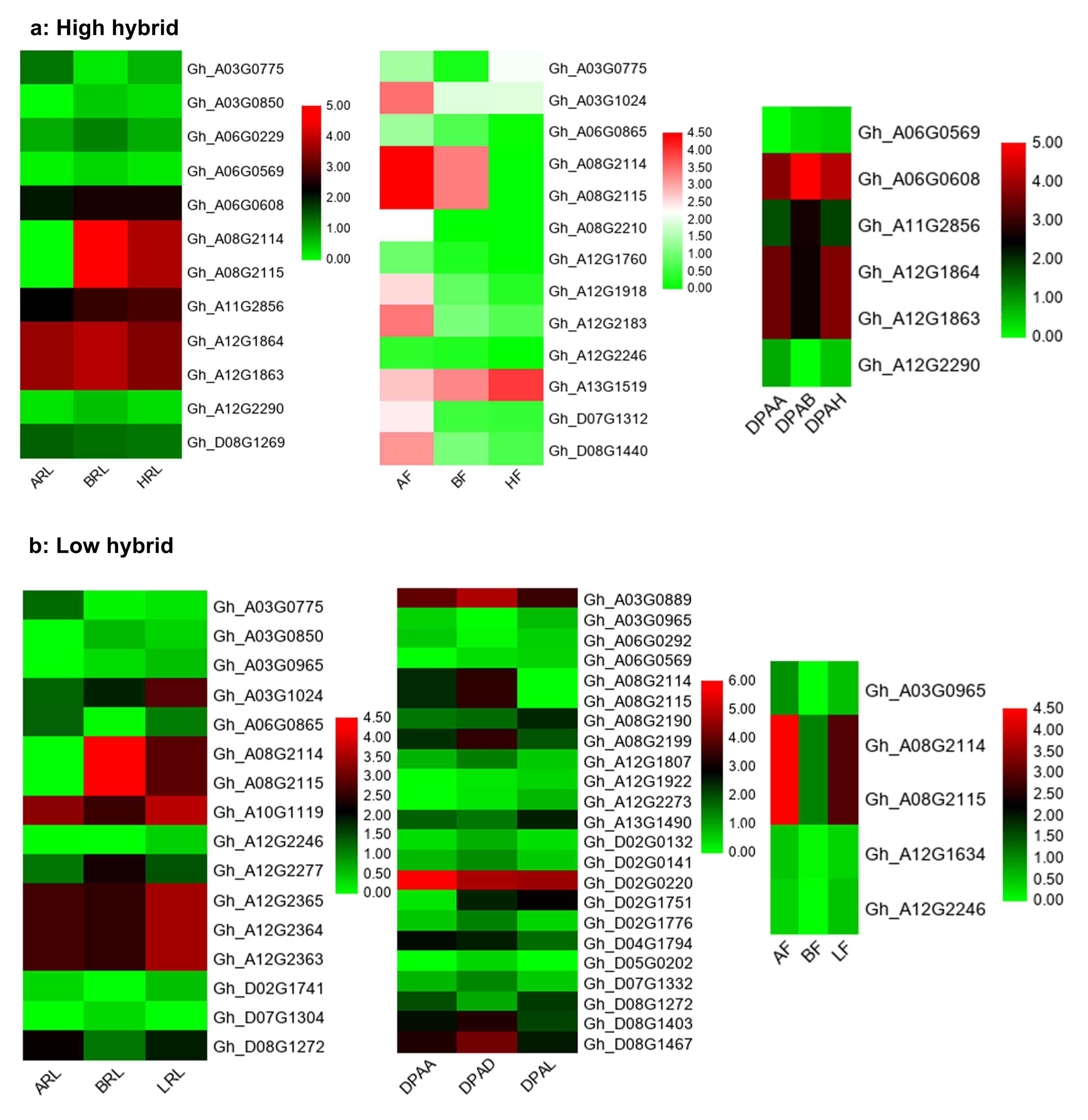
To construct the significant regulatory network for yield heterosis, first, we selected blue gene module in both hybrids. Secondly, the DEGs mapped on SCY-QTLs together with their interaction genes having weight ≥0.40 were screened (Table S6). Subsequently, our constructed gene co-expression network showed that nine SCY-QTLs genes had many interconnected genes involved in biological and molecular functions such as cell wall regulation or modification, carbohydrate metabolic processes, ion binding and transportation, biological regulation, methylation, endoplasmic reticulum, actin filament processing, catalytic activities, and protein binding or transportation (Fig. 9). We considered these nine genes as putative candidate genes for yield heterosis. Out of nine genes, two genes Gh_A03G1024 (BZR1: Brassinazole-resistant 1) and Gh_D08G1440 (ASK8: Shaggy-related protein kinase theta) involved in plant hormone signal transduction pathway showed differential expressions in flower buds of high hybrid. Another three starch and sucrose metabolism pathway also had differential expressions in flower buds of high hybrid. These genes were Gh_A08G2210 (At3g43860: Endoglucanase 16), Gh_A12G2183 (GBSS1: Granule-bound starch synthase 1, chloroplastic/amyloplastic), and Gh_D07G1312 (APL2: Glucose-1-phosphate adenylyltransferase large subunit 2, chloroplastic). The remaining four had specific differential expression in 1DPA ovule of low hybrid. Three of them denoted as Gh_D08G1467 (MPK4: Mitogen-activated protein kinase 4), Gh_A03G0889 (PHO1: Phosphate transporter PHO1 homolog 3), and Gh_A08G2199 (JAZ10: Jasmonate-zim-domain protein 10) were enriched in plant hormone signal pathway. The remained one gene Gh_D05G0202 (CRR21: Pentatricopeptide repeat-containing protein At5g55740, chloroplastic) belonged to starch and sucrose metabolism pathway. Collectively, gene co-expression network analysis in hybrids not only helped to screen key genes but also provides novel insights into the regulatory mechanism of yield heterosis of cotton.
Quantitative real-time PCR validation of RNA-Seq data
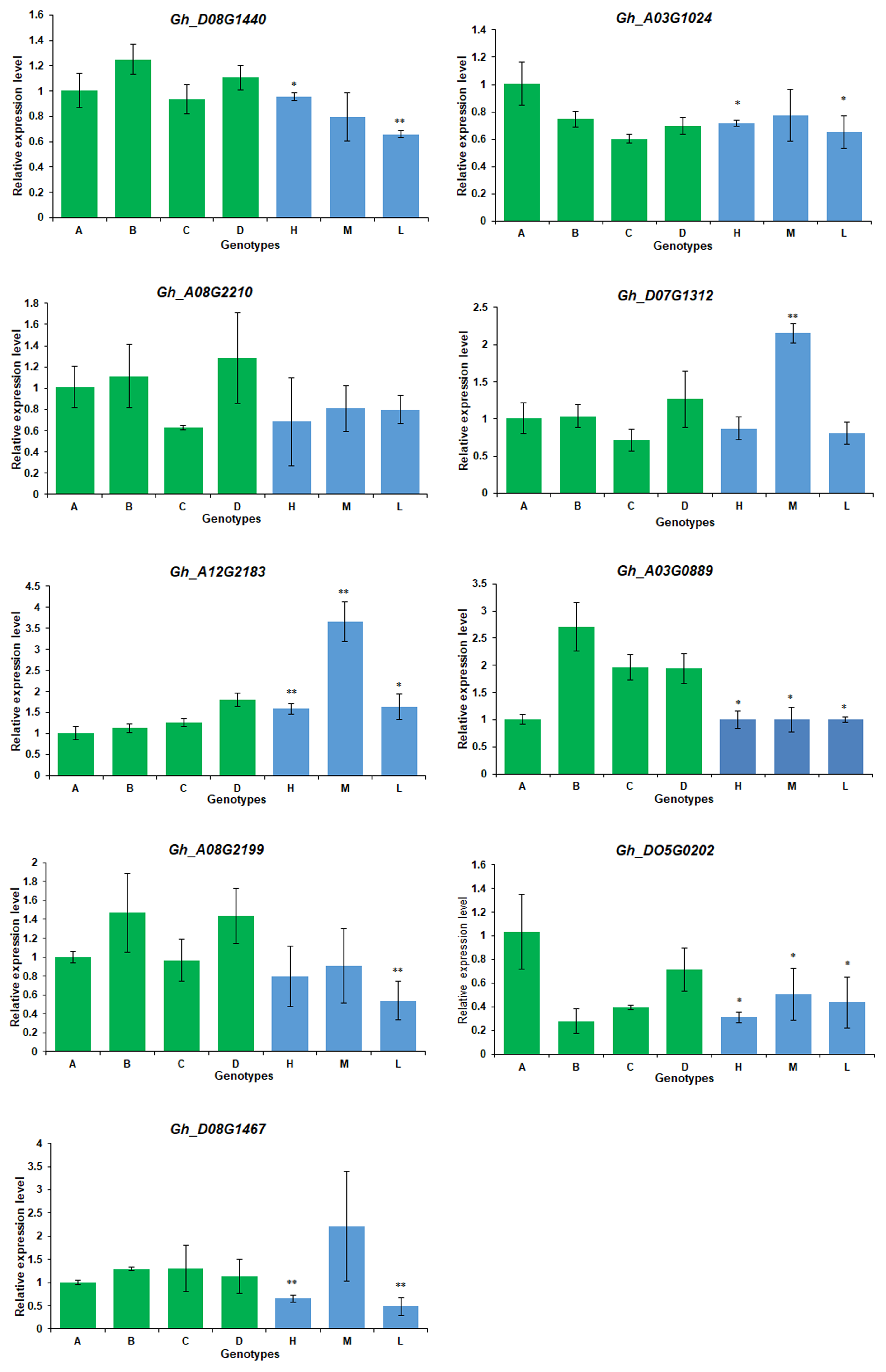
Nine DEGs were selected to perform real-time qRT- PCR analysis. These genes showed hybrid and tissue-specific expression in RNA-seq, mapped on SCY-QTLs, and had the highest interconnected genes in the co-expression network. Primer was designed in gene-specific way to ensure accuracy of expression (Table S7). Analysis results showed Gh_A03G1024, and Gh_D08G1440, Gh_A12G2183, Gh_A03G0889, Gh_D05G0202, Gh_A08G2199, Gh_D08G1467 and Gh_D07G1312 displayed a significant change in each hybrid compared from their one parent or both parents (Fig. S11). However, gene Gh_A08G2210 showed no statistical difference in hybrids. More interestingly, these genes apparently showed the expression similar to their maternal or paternal parent as determined in RNA-seq results.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}