Reagent
Internal standards, including lysophosphatidylcholine C19:0 (LPC 19:0), phosphatidylcholine C38:0 (PC 38:0), phosphatidylethanolamine C30:0 (PE 30:0), sphingomyelin C12:0 (SM 12:0), ceramide C17:0 (Cer 17:0), fatty acid C16:0-d3 (FA 16:0-d3), fatty acid C18:0-d3 (FA 18:0-d3) and triglyceride 45:0 (TG 45:0) were purchased from Avanti Polar Lipids (Alabaster, AL). Internal standards, including acetylcarnitine-d3 (carnitine C2:0-d3), carnitine C10:0-d3, carnitine C16:0-d3, tryptophan-d5, phenylalanine-d5, cholic acid-d4, chenodeoxycholic acid-d4) were purchased from Sigma company (US). Ammonium acetate and tert-butyl methyl ether (MTBE) were purchased from Sigma company (US). Acetonitrile, methanol and isopropanol were purchased from Merk company (German). Milli-Q water was made from a Milli-Q system (Millipore,Billerica,MA). Pyridine, methoxyamine hydrochloride, and N-Methyl-N-(trimethylsilyl) trifluoroacetamide (MSTFA) for GC-MS derivatization were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Sample preparation
For GC-MS based metabolomics. The serum samples were thawed at room temperature and vortex for 5 sec. Then 50µL serum sample was drawn into to 1.5 ml eppendorf tubes, and 200µL methanol containing internal standard (10 µg/mL tridecanoic acid) was added subsequently for metabolite extraction and protein precipitation. After full votex for another 5 min at room temperature, the supernatant was removed and lyophilized in new 1.5 ml eppendorf tubes. Each aliquot was reconstituted in 50 µl methoxyamine pyridine solution (20 mg/mL). After full vortex, the tubes with the solutions were incubated in water bath at 37 ℃ for 1.5 h. Then, silylation reaction lasted for 1 h at 37 ℃ in water bath after adding 40 µl of MSTFA. The tubes were centrifuged at 13 000 g for 10 min at 4 ℃, and the supernatants were drawn for GC-MS injections.
For LC-MS based metabolomics. A 96-well filter protein precipitation plate was used for sample preparation. An aliquot of 50 µL serum was drawn into each well, and 200 µL extraction solvent was added subsequently for metabolite extraction and protein precipitation. This extraction solvent was methanol containing internal standards (0.5 ug/mL fatty acid (FFA) C16:0-d3, 0.5 ug/mL FFA C18:0-d3, 0.16 ug/mL carnitine C2:0-d3, 0.1 ug/mL carnitine C10:0-d3, 0.075 ug/mL ug/mL carnitine C16:0-d3, 0.75 ug/mL LPC 19:0, 4.25 ug/mL tryptophan-d5, 3.61 ug/mL phenylalanine-d5, 0.37 ug/mL cholic acid-d4, 0.3 ug/mL chenodeoxycholic acid-d4). Then the plate was covered with aluminum foil and vortexed for 10 min, then it was centrifuged for collecting supernatant. The extracts were lyophilized and stored at -80 ℃ before LC-MS analysis.
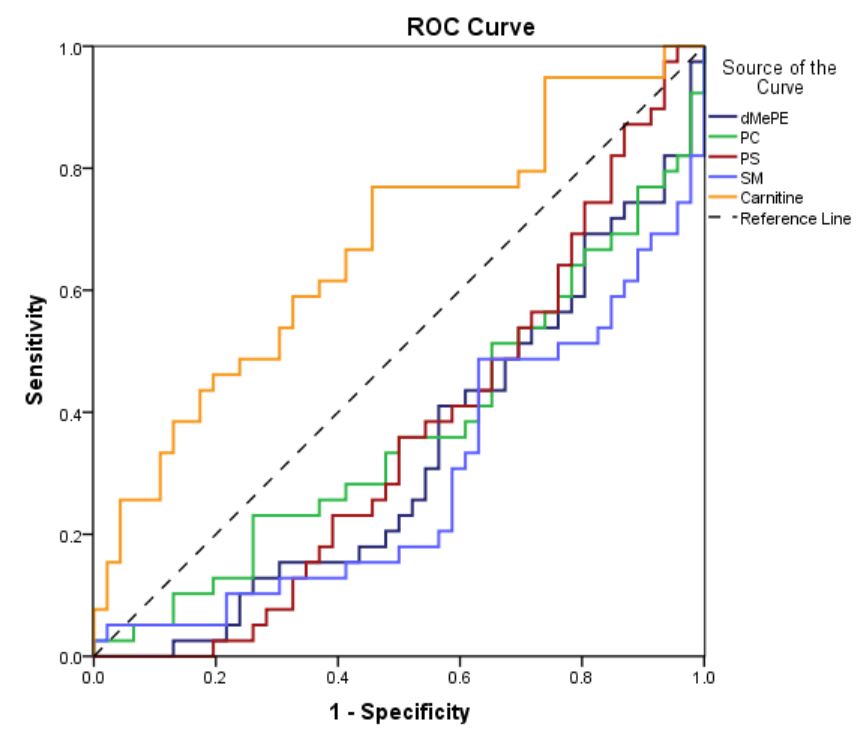
For LC-MS based lipidomics. An aliquot of 40 µL of serum for each sample was drawn into eppendorf tube. And 300 µL of methanol containing LPC 19:0 (0.80 µg/mL), PC 38:0 (1.00 µg/mL), PE 30:0 (0.75 µg/mL), SM 12:0 (0.65 µg/mL), Cer 17:0 (0.80 µg/mL), FFA 16:0-d3 (0.50 µg/mL), FFA18:0-d3 (0.20 µg/mL), TG 45:0 (0.60 µg/mL) was added, followed by vortex mixing for 30 sec and protein denaturation. Then, 1 mL of MTBE and 250 µL of water were added into the mixture followed by vortex mixing for 30 sec to extract lipid compounds. After centrifugation at 14000 rpm at 10 ℃ for 10 min, 400 µL of organic supernatant was collected and lyophilized, then stored at -80 ℃.
Data acquisition.
For GC-MS based metabolomics. One microliter of the above derivatized products were injected into a gas chromatograph coupled to a plus quadrupole mass spectrometer detector (GCMS 5977A; Agilent, US). The split ratio of 1:10 was employed. A DB-5 MS capillary column (length 30 m x ID 0.25 mm, film thickness 0.25 µm) (J&W Scientific, Folsom, CA, USA) was employed for metabolite separation. The flow rate of the helium gas was 1 mL/min; the oven temperature began at 80 ℃ and maintained for 1 min, which was linearly increased to 210 ℃ at 30℃/min, and then linearly increased to 320 ℃ at 20 ℃/min, and holding for 4 min at 320 ℃. The temperature of the ion source was 230 ℃. The data was acquired in full scan mode with the scan range set at 33–600 Dalton.
For LC-MS based metabolomics. The dried powder was reconstituted in 80 µL acetonitrile /water (1:4) and centrifuged at 13 000 g for 10 min at 4 ℃. 5 µl of the supernatant was injected into a Vanquish UPLC- Q Exactive (Thermo Fisher Scientific, Rockford, IL, USA) system. For positive ionization mode, a 50 mm × 2.1 mm, 1.7 µm Waters BEH C8 (Waters, Milford, MA) column was used for separation; Mobile phase A was 0.1% formic acid in water and mobile phase B was 0.1% formic acid in acetonitrile. The elution gradient program started from 5% B and kept for 0.5 min, then linearly increased to 40% B within 1.5 min, continued to 100% B in 6 min and maintained at 100% B for 2 min, then back to 5% B within 0.1 min. A post- equilibration was kept for 2.5 min. For negative ionization mode A, a 50 mm × 2.1 mm, 1.8 µm ACQUITY UPLC HSS T3 (Waters, Milford, MA) column was used for separation. Mobile phase A was 6.5 mM ammonium bicarbonate in water and mobile phase B was 6.5 mM ammonium bicarbonate in 95% methanol/water. The elution gradient program started from 2% B and kept for 0.5 min, then linearly increased to 40% B within 2 min, and further increased to 100% B within another 6 min and maintained at 100% B for 2 min, then back to 2 %B in 0.1 min. A post-equilibration was maintained for 2 min. In both positive and negative ionization mode, the oven temperature was 60 ℃ and the flow rate was 0.4 mL/min. For MS detection, resolution was set at 120 K and full scan mode was employed with m/z scan range 80-1200. The spray voltage was 3.5 kV for positive ionization mode and 3.0 kV for negative ionization mode, respectively. The capillary temperature was 300 ℃ and auxiliary gas heater temperature was set at 350 ℃. The flow rates of sheath gas and auxiliary gas were set at 45 and 10 in arbitrary units.
For LC-MS lipidomics. The dried sample was reconstituted in 30 µL chloroform/ methonal (v/ v, 2:1) and 60 µL acetonitrile/isopropanol/MilliQ water (v/ v/ v, 65: 30: 5). An aliquot of 5 µL reconstituted solution was injected into a Waters UPLC system (ThermoFisher, US) coupled to a Q ExactiveTM HF (ThermoFisher, US). A ACQUITY UPLC C8 column (100 mm × 2.1 mm × 1.7 µm) was used for separation. Mobile phase A was 60% acetonitrile in water containing 10 mM ammonium acetate. Mobile phase B was 10% acetonitrile in isopropanol containing 10 mM ammonium acetate. Elution gradient started with 32% B and kept for 1.5 min. Then it was linearly up to 85% within next 14 min, then to 97% B within 0.1 min and kept for 2.4 min. The gradient was back to 32% B within next 0.1 min and kept for 1.9 min. Flow rate was set at 0.26 mL/min. Column temperature was 55 ℃. For MS detection, resolution was set up 120 K and full scan mode was employed. The scan range was 200–1100 Da for positive ionization mode, 120–1600 Da for negative ionization mode. The spray voltage was 3.5 kV for positive ionization mode and 3.0 kV for negative ionization mode, respectively. The capillary temperature was 300 ℃and auxiliary gas heater temperature was set at 350 ℃. The flow rates of sheath gas and auxiliary gas were set at 45 and 10 in arbitrary units.
Data preprocessing.
For GC-MS metabolomics data preprocessing, to increase the detection of the low-abundance metabolites, two-fold QC samples was used for metabolite identification via library searching (NIST). Both comparison of mass spectrum similarity and retention index distance of reference standards in our in-house library were employed for metabolite identification. Then, the identified table was used for the next peak integration by MassHunter Workstation software (Agilent, US).
For LC-MS metabolomics data preprocessing, after automated peak detection, alignment, and integration, peaks were identified according to our in-house database containing more than 2,000 metabolite standards.
For lipidomics data preprocessing, lipid identification was performed followed by peak integration for individual lipid molecule. Lipid SearchTM 4.1 (ThermoFisher, US) was employed to obtain general information of lipid candidates, including m/z, retention time, (sub)class, feature fragments and mass accuracy (in ppm). Then, manual check d for detailed structural information and known peak extraction were performed. The details in annotation can be referred to our previous publication[11].
Mass spectrometry responses of metabolites or lipids were normalized to internal standards. For LC-MS metabolomics and lipidomics analyses, internal standards were chosen firstly according to their corresponding classes. If no, then internal standards were chosen according to similarity in structure or retention time.
{kind=link}