Animals
Wild-type adult female NMRI mice (6–8 weeks old) were purchased from Pasteur Institute (Tehran, Iran). The female mice were used for ovarian cell isolation of ovarian tissues. The nude female mice that were used in the transplantation experiments and teratoma assessments were obtained from Pasteur Institute (Tehran, Iran). All animals were maintained on a 12 h light-dark cycle with access to water and standard chow ad libitum. The Royan Institutional Review Board and Institutional Ethical Committee of Royan Institute approved all animal care studies and procedures (No: Ec/92/1026).
Mice ovarian cell isolation
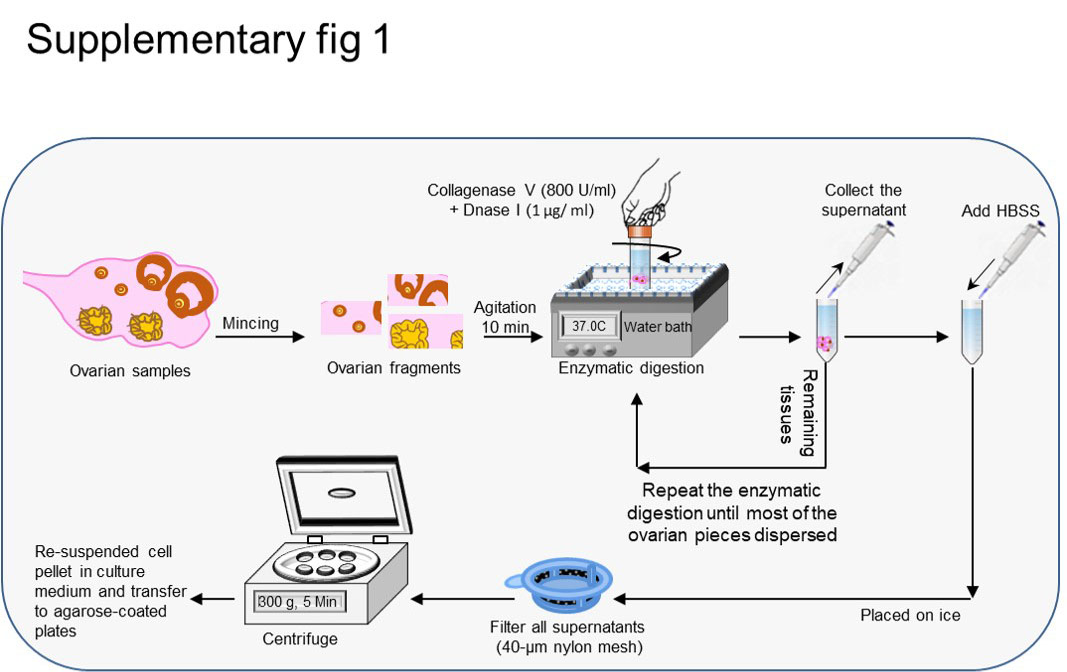
For each experiment, we collected ovarian tissues from 4 mice (8 ovaries). The fat pad, bursa, and oviduct were carefully removed from each of the ovarian tissues. Then, the ovaries were minced and washed twice in Hank’s balanced salt solution (HBSS, Sigma-Aldrich) that contained 1% penicillin and streptomycin (Invitrogen). Ovarian tissues underwent enzymatic digestion along with a gentle hand orbital shaking at 37°C in a water bath for 10 min in a solution of 2 ml HBSS that included 800 U/ml of type IV collagenase (Invitrogen) and 1 μg/ ml DNase I (Sigma-Aldrich). Because of the influence of mechanical stress on cell viability during isolation (26), we did not use from orbital shaker for ovarian cell dissociation, that led to increased cell viability and later aggregate formation (data not shown). Moreover, stem cells are sensitive to enzymes (27), therefore, fresh enzyme solution was prepared for each experiment. We replaced 1.5 ml of the supernatant with 1.5 ml fresh enzyme solution and continued the procedure until the majority of the ovarian tissues were dissociated (Supplementary Fig. 1). The collected supernatants were kept on ice for each repeat of the procedure. Finally, the supernatants collected from all of the procedures were filtered through a 40 μm nylon mesh and centrifuged at 300 g for 5 min, then washed twice with HBSS. The supernatant was removed carefully and the clumps of cells were resuspended in culture medium.
Human ovarian tissues
Human ovarian tissue samples were obtained after provision of written informed consent from eight reproductive age women (35–45 years of age) who presented for treatment of uterine disorders. These women were candidates for either a myomectomy or hysterectomy due to various reasons that included uterine fibroids which caused pain and bleeding or they were candidates for tubectomy for blocked fallopian tubes. Each woman signed an informed consent for participation and a questionnaire that recorded sex, age, and medical history was completed at the time of enrollment. The Royan Institutional Review Board and Institutional Ethical Committee of Royan Institute (No: Ec/92/1026) approved the use and preparation of the human ovarian samples for this study.
Human ovarian cell isolation
Biopsies of the human ovary tissues (2×2×2 cm) were transported in α-MEM media that contained penicillin (100 U/ml) and streptomycin (100 μg/ml) (both from Invitrogen) on ice to the laboratory. The biopsies were gently washed several times in HBSS that contained antibiotics. The ovarian tissues were dissected and minced by a sterile surgical instrument and rinsed with HBSS. Tissue dissociation was performed by using the enzymatic digestion method described for mice ovaries.
Aggregate formation
We seeded the single cells on agarose-coated plates to generate ovarian aggregates from the ovarian cells. For optimization of cell seeding, we used 1×105–5×105 cells per 3 cm2 plate in 2 ml medium and observed that 3×105 cells/3 cm2 plate (Falcon) were suitable for aggregate formation over 2–3 days of culture (data not shown). Seeding less than 3×105 cells decreased both the size and number of the aggregates, and resulted in their gradual degeneration. Aggregate formation with higher numbers of cultured cells joined together after 24 h and formed more dense aggregates. The cells were seeded on plates precoated with 1% agarose and incubated at 37°C and 5% CO2 in α-MEM (Invitrogen) supplemented with 10% FBS (Hyclone), 1 mM sodium pyruvate (Invitrogen), 1 mM nonessential amino acids (Invitrogen), 2 mM L‑glutamine (Sigma), 1X penicillin-streptomycin (Invitrogen), 0.1 mM β-mercaptoethanol (Sigma-Aldrich), 1% N2 supplement (R&D), 103 units/ml leukemia inhibitory factor (LIF, Royan BioTech), 10 ng/ml recombinant human epidermal growth factor (rhEGF, Royan BioTech), 1 ng/ml basic fibroblast growth factor (bFGF, Royan BioTech), and 40 ng/ml glial cell-derived neurotropic factor (GDNF, Royan BioTech). After 2–3 days of cell culture, we observed the formation of round or oval aggregates of ovarian cells that were 50–100 µm in diameter.
In vitro derivation and expansion of ASCs
The aggregates were collected using a thin Pasteur pipette and plated in 12-well plates (approximately 5 aggregates per well). The 12-well plates were precoated with mitotically inactivated mouse embryonic fibroblasts (MEF), which were treated with mitomycin (Sigma-Aldrich). After three days, we observed the appearance of round cells that migrated from the aggregates and expanded in the culture. These cells were similar in size and morphology to the previously reported germline stem cells by White et al.(9).
The ASCs proliferated in culture over time. The medium was renewed every 2–3 days. The mice ASCs were passaged initially at a 1:2 split ratio with 0.05% trypsin-EDTA (Invitrogen) and re-plated on fresh MEF; after 4–5 passages, they were split at a 1:3 ratio. The human ASCs were passaged every 30 days with a split ratio of 1:2.
In vitro differentiation of ASCs into oocyte-like cells
Spherical oocyte-like cells (OLCs) spontaneously formed in culture during in vitro expansion of the ASCs. For directed oocyte differentiation, we cultured the ASCs in differentiation medium that contained α-MEM supplemented with 10% FBS, 1 mM sodium pyruvate, 1 mM nonessential amino acids, 2 mM L‑glutamine, 1X concentrated penicillin-streptomycin, 0.1 mM β-mercaptoethanol, 1% N2 supplement (R&D), 10 ng/ml rhEGF, 5 μl/ml insulin/transferrin/selenium (Invitrogen), 0.05 IU follicle stimulating hormone (FSH; Sigma-Aldrich), and 0.03 IU luteinizing hormone (LH; Sigma-Aldrich). Each 2–3 days we replaced half of the medium with fresh medium. Supplementary Material Table S3 lists the differentiation media.
ASCs cryopreservation
The ASCs were trypsinized and gently mixed with cryoprotective solution (90% FBS and 10% DMSO), incubated overnight at -80°C, and then transferred to liquid nitrogen.
Flow cytometry
We performed flow cytometry analysis to assess the effects of cryopreservation on ASCs viability. Propidium iodide (PI) was used to detect the nonviable cells. Cells before and after cryopreservation were incubated with 0.05% trypsin-EDTA at 37°C for 3 min to obtain a single-cell suspension. We added PI to the cell suspension just prior to FACS analysis to enable detection of any nonviable cells by the BD FACS Aria II.
Reverse transcription-polymerase chain reaction (RT-PCR) analyses
Total RNA was extracted with TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. Reverse transcription was performed using the cDNA Synthesis Kit (K1632, Fermentas). The presence of each indicated mRNA was analyzed by conventional RT-PCR using the primers listed in Table S1.
Measurements of 17 β-estradiol and inhibin
We evaluated the endocrine functions of ovarian cells in the aggregates by measuring the levels of 17 β-estradiol (E2) and inhibin in the culture media. Ovarian cells were cultured in the presence and absence of 0.01 IU FSH and 7.5 IU LH for two days under 3D culture conditions. The culture media was collected to assess the secretion of these hormones with an ELISA kit (Shanghai Crystal Day Biotech Co., Ltd.) according to the manufacturer’s instructions.
Alkaline phosphatase
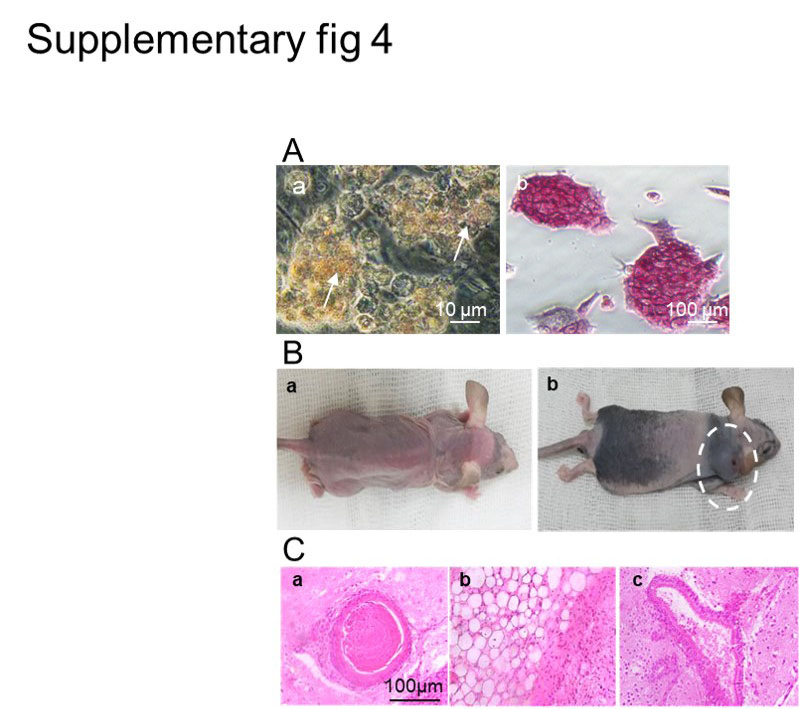
We used an Alkaline Phosphatase Staining Kit (Sigma-Aldrich) to detect alkaline phosphatase (AP) activity in the ASCs. Mouse embryonic stem cells (mESCs) were used as the positive control.
Immunocytofluorescence staining
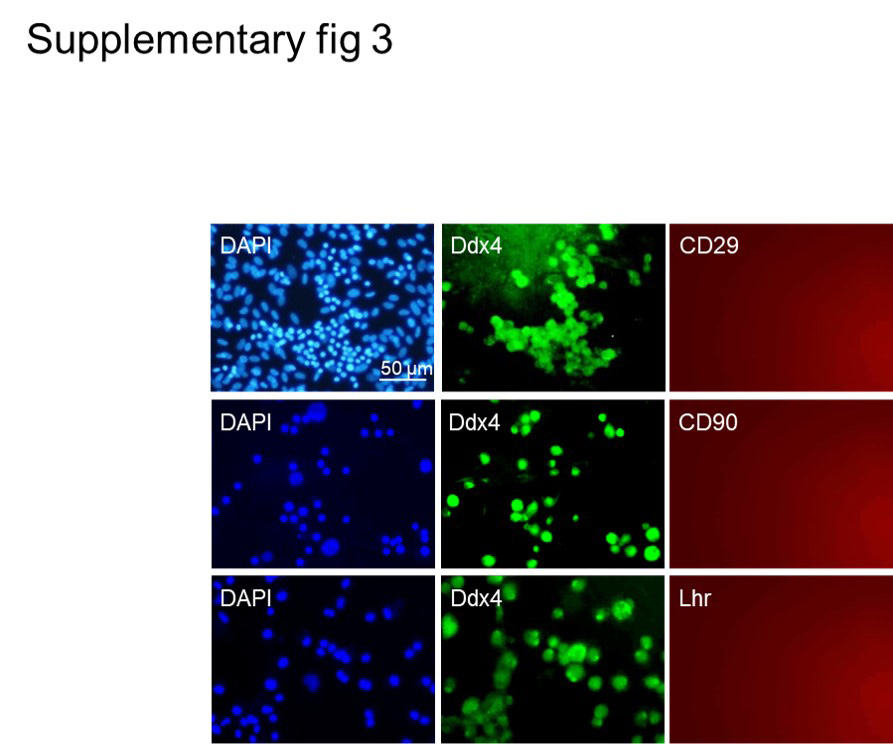
aggregate-derived cells were washed with 1X-concentrated phosphate-buffered saline (PBS) and fixed in 4% PFA for 20 min. After permeabilization with 0.5% Triton X-100, the cells were incubated for 1 h in blocking buffer that consisted of PBS and 10% normal goat serum, followed by an additional overnight incubation period with primary antibody (Table S2) in a humidified chamber at 4°C. The cells were subsequently incubated with the appropriate secondary antibody at room temperature for 45 min. For nucleus staining, we used 4,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich). Immunostaining without primary antibodies was used as the negative control for the cells.
Histological analysis
Teratomas in the teratoma assessment groups and ovarian tissue in the Sterilized mice were fixed in 10% formalin and 4% Bouin’s solution. They were subsequently dehydrated in a graded ethanol series, vitrified in xylene, and embedded in paraffin and finally sectioned into 6 μm sections. These sections were mounted onto slides. The slides were de-waxed in xylene and rehydrated through an ethanol gradient and stained with hematoxylin and eosin (H&E, Sigma-Aldrich).
Detection of apoptosis by the TUNEL assay
We used the TUNEL assay to evaluate cell apoptosis in the aggregates. The aggreagte sections were each washed twice in PBS for 5 min after deparaffinization. The sections were permeabilized by incubation in 0.1% Triton X-100 and 0.1% sodium citrate for 8 min at room temperature. The sections were washed twice with PBS and incubated with the TUNEL reaction mixture (5 µl enzyme solution and 45 µl labeling solution) for 60 min at 37ºC in the dark. The negative control consisted of tissue sections incubated with only labeling solution. After incubation with the reaction mixture, the sections were washed three times with PBS. Apoptotic cells (green fluorescence) were visualized by a fluorescence microscope (Olympus). Counterstaining with DAPI was used to visualize the nuclei.
This assay was performed using the Death Detection Kit (Roche Molecular Biology, Germany), which identifies apoptotic fragmented DNA by terminal deoxynucleotidyl transferase (TdT)-mediated incorporation of fluorescein-12-dUTP into the 30-hydroxyl end of DNA TdT.
Teratoma formation
We subcutaneously injected 1 x 105 ASCs into the neck area of three nude female mice (Pasteur Institute, Tehran, Iran). Controls were age-matched female nude mice that were injected at the same site with mouse ESCs (mESCs). The mice were monitored weekly for teratoma formation for up to three months.
Sterilization of adult mouse ovaries by chemotherapy
We intraperitoneally injected busulfan (30 mg/kg, resuspended in DMSO) and cyclophosphamide (120 mg/kg) into 2-month-old C57BL/6 female wild-type mice (28) (n=10). Controls were injected with DMSO. The transplantation of ovarian cells was performed one week after chemotherapy treatment and assessments were done 4 weeks after cell transplantation.
Sample preparation for RNA sequencing analysis
To assess gene expression profiles throughout the course of aggregate cell derivation, we initially collected samples from aggregates, ASCs at different time points (primary culture: P0, 4, 10, and 40) and MEFs (at least three biological replicates in each group). These time points were chosen to evaluate changes in transcriptions that occurred during derivation of the aggregate cells and their long-term culture. Due to the presence of MEF cells with different densities during the culture of the aggregate-derived cells, we chose the MEF samples for this cell contamination. The cells were collected and preserved at -80°C until RNA extraction.
Analysis of RNA-sequencing data
Initially all reads were controlled by FastQC (version 0.11.7)(29) followed by Trimmomatic (version 0.38)(30) for trimming low quality bases. The remaining reads were aligned to the mouse reference genome GRCm38 (downloaded from Ensembl database) (31) using HISATt2 (32). In order to find the differentially expressed genes (DEGs), we used the HTSeq (version 0.9.1)(33) and DESeq2(34) packages and the R program for finding clusters using the K-means algorithm. We performed gene ontology (GO) analysis using the Enrichr (35) package in the R program for both enriched GOs and biological pathways.
Heat map clustering, volcano plot, principal component analysis (PCA), and K-means clustering were performed using the R studio platform. GO and functional enrichment were assessed with Enrichr (https://amp.pharm.mssm.edu/Enrichr/) by taking into consideration enriched functions in at least two different data bases (Kyoto Encyclopedia of Genes and Genomes [KEGG] 2016, Wiki Pathways 2016, Biocarta 2016, and Reactome Pathway 2016 as well as GO terms 2018).
Small molecule treatment
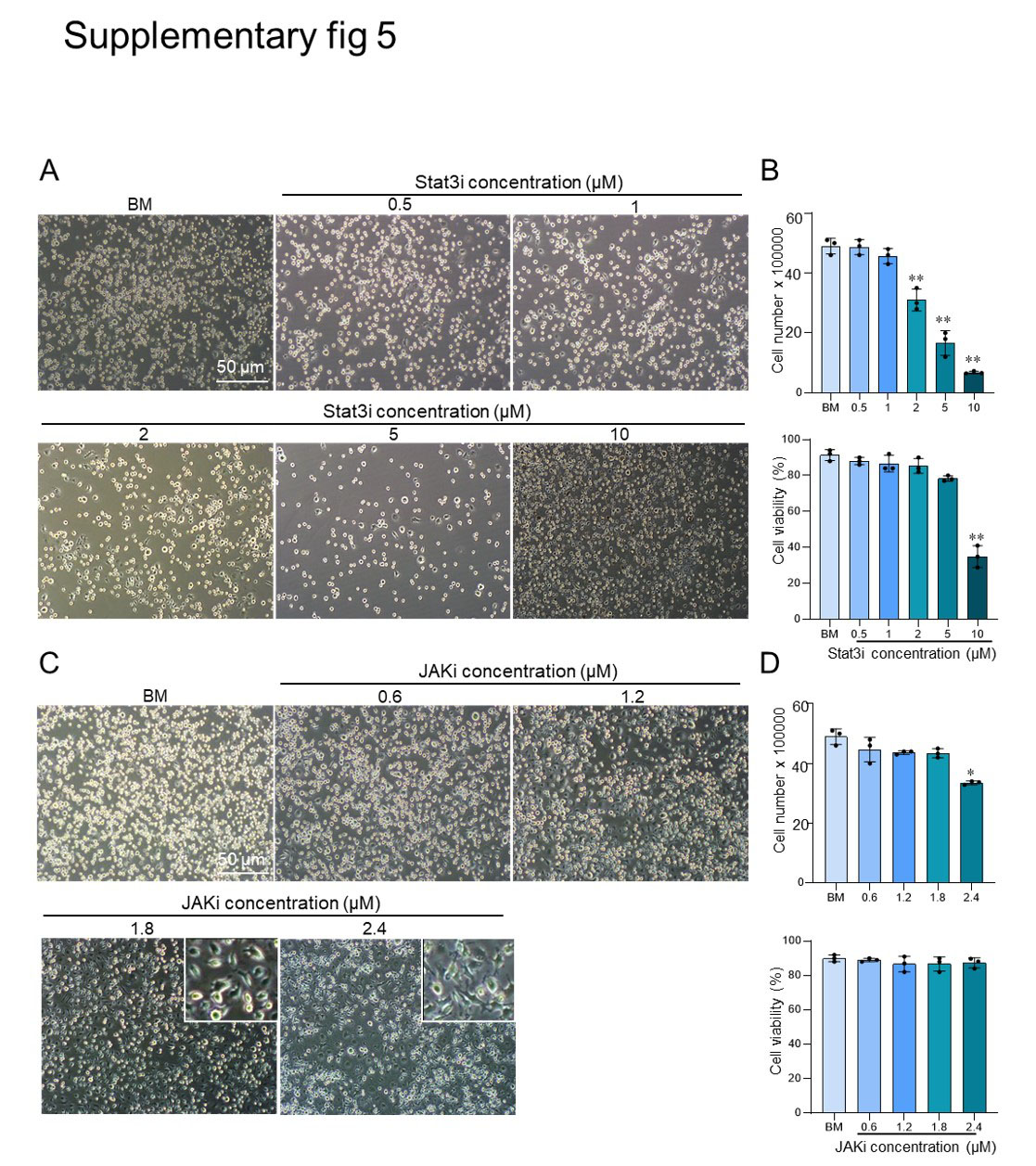
We plated 200.000 aggregate-derived cells that were cultured for at least 40 passages onto 12-well plates with basal culture medium or basal medium supplemented with 10 ng/ml IL-6 (36), different concentrations of JAK inhibitor and Stat3 inhibitor or basal medium without LIF. Three replicates for each condition were examined. Cell number and viability were counted after three days using a hemocytometer after trypan blue staining.
Image analysis and cell counts
We determined the numbers of ASCs, OLCs, and Ki67-positive cells with wide angle (20x) images that covered approximately 200–1000 cells per image in ImageJ software using the ‘cell counter’ plugin. We counted at least five fields of view for every condition.
Statistical analysis
All experiments were conducted in at least three independent replications. All data are expressed as mean ± SD, and analyzed by student’s t-test. All graphs were generated using GraphPad Prism software version 8.0. P-values <0.05 were considered significant.
Data availability
The datasets and computer code produced in this study are available in the following database:
RNA-Seq data: Gene Expression Omnibus (GEO) under accession number the submission ID= PRJNA664684.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}