Previous studies in our laboratory have shown that LC18002 does not contain mutations in the direct target of cyhalofop-butyl, implying that its resistance is most likely operating via an NTSR mechanism. Metabolism-based resistance to herbicides that inhibit ACCases has been reported extensively, with instances increasing more in recent years [14]. RNA-seq has been successfully used to identify genes involved in metabolic resistance to acetolactate synthase (ALS) herbicides in two grass weed species, including Wimmera ryegrass (Lolium rigidum Gaud.) [15] and slender foxtail (Alopecurus myosuroides) [16]. However, no study has yet been conducted at the whole transcriptome level to understand potential NTSR mechanisms involved in L. chinensis resistance. Identifying genes involved in NTSR is crucial to better understand the evolution of metabolic resistance, which may lead to improved weed management strategies.
Transcriptome sequencing and screening
RNA obtained from the different L. chinensis samples was paired-end sequenced using an Illumina HiSeq2500. The raw reads were then trimmed to remove adaptors, repetitive sequences and low-quality reads (Table 1). The average GC content was about 53%, Q20 value was above 96%, Q30 value was above 89%, and the Q30 value of 95% of all samples was above 91%. Clean reads were then mapped against the reference genome using Hisat2. Mapping rate, number of clean reads, clean bases, and additional statistics are shown in Table 1. In addition, we also analyzed the FPKM distribution of each sample, and the results showed that the distribution of FPKM was uniform in each sample, which was suitable for subsequent analysis (Additional file 6).
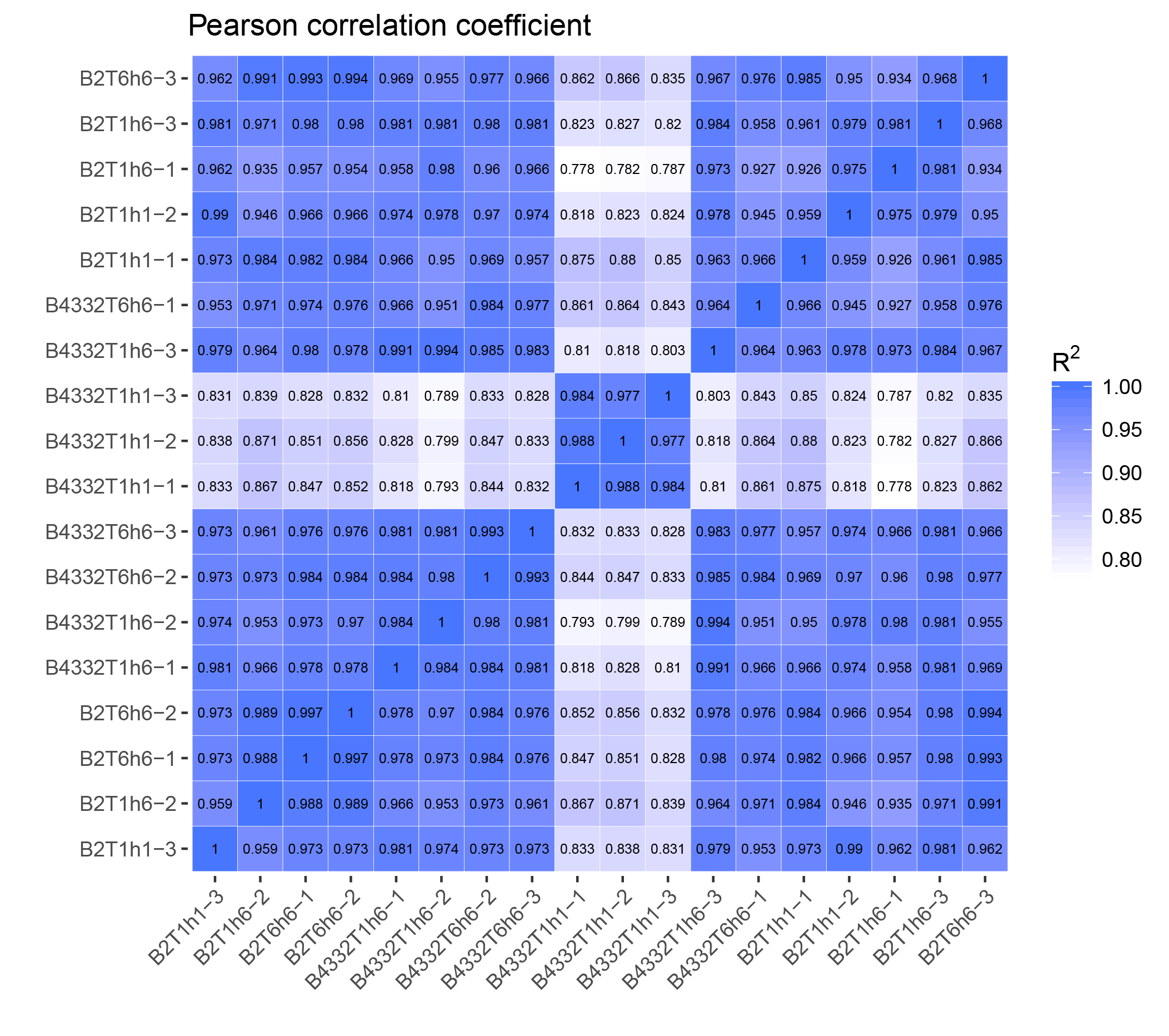
We next employed correlation analysis to determine whether replicates had similar expression values, which is a well-established method for assessing data quality. Our analysis revealed that the majority of replicates had correlation coefficients of 0.97 or higher (Additional file 1). Additionally, the correlation coefficient between B4332T1h1 and B2T1h1 was only 0.83, indicating a large difference between the two materials.
Identification of transcription factors and differential gene expression
Transcription factors are a group of protein that can bind specifically to a specific sequence at the upstream of a gene, so as to ensure the expression of the target gene at a specific time and space with a specific intensity. Transcription factor prediction was analyzed by iTAK software and HMMSCAN was used to identify TF, Transcription factor was compared to plant specific transcription factor database PlnTFDB. A total of 2,876 transcription factors (TFs) and transcriptional regulators from the transcriptome were identified (Table 2). TFs form complex regulatory networks that control the expression of their target genes. These regulatory circuits are pivotal for coordinating transcriptional and post-transcriptional control of target genes [17]. SNPs in TFs have previously been shown to impact agriculturally important traits. For example, a negative regulator of DELLA genes has been shown to cause dwarfism in oil plants [18]. In Prunus, TFs control many agriculturally important traits such as the flowering, fruit quality, and biotic and abiotic stress resistance [19].
EdgeR software was then used to analyze the differential expression of genes in each sample, and the corrected p-value and PADj value of the differential expression tests were calculated. The input data for differential expression analysis consisted of read counts obtained via quantitative analysis. Detailed label names and their treatment condition are shown in Additional file 8. The number of differentially expressed genes in each treatment is shown in Table 3.
As shown in Figure 1, there were 150 differentially expressed genes (DEGs) in A2 versus A3, among which 96 genes were up-regulated and 54 genes were down-regulated. There were 749 DEGs in A1 versus A2, among which 137 genes were up-regulated and 612 genes were down-regulated. There were 1753 DEGs in A1 versus A3, among which 678 genes were up-regulated and 1075 genes were down-regulated. There were a total of 14709 DEGs in B1 versus A1, among which 6147 genes were up-regulated and 8562 genes were down-regulated. There were 1649 DEGs in B2 versus B3, among which 658 genes were up-regulated and 991 genes were down-regulated. There were 17286 DEGs in B1 versus B2, among which 7275 genes were up-regulated and 10008 genes were down-regulated. There were 15194 DEGs in B1 versus B3, among which 6357 genes were up-regulated and 8837 genes were down-regulated. There were a total of 683 DEGs in B2 versus A2, among which 237 genes were up-regulated and 446 genes were down-regulated. There were a total of 2704 DEGs in B3 versus A3, among which 2065 genes were up-regulated and 639 genes were down-regulated.
In order to further screen candidate herbicide genes, we believe that the following 6 contrast groups are more suitable for screening. B1 versus A1 was used to compare the difference between the sensitive and resistant material, while B3 versus A3 was used to compare the different results of cyhalofop-butyl treatment in the two L. chinensis populations. B2 versus B3 was used to measure the gene expression changes in sensitive L. chinensis genes caused by cyhalofop-butyl treatment. A2 versus A3 was used to determine which genes were induced by cyhalofop-butyl treatment in resistant L. chinensis genes. The four sets of DEGs were plotted on a Venn diagram (Figure 2). A total of 12829 DEGs were found exclusively in the A1 versus B1 group, indicating that the two materials were significantly different even without cyhalofop-butyl treatment. Additionally, 1426 DEGs were found uniquely in the B3 versus A3 comparison, meaning these genes were only different during treatment with cyhalofop-butyl. These genes therefore represent possible candidates for the NTSR present in the resistant L. Chinensis line. Another 506 genes were found to be differentially expressed only in the B2 versus B3 comparison group, indicating that they represent a response present only in L. chinensis that is sensitive to cyhalofop-butyl and only 28 DEGs existed exclusively in the A2 versus A3 comparison, indicating that they represent a response present only in L. chinensis that is risistant to cyhalofop-butyl In addition, the B1 versus B2 comparison was used to determine the effect of water treatment on the expression of rice genes in the sensitive population, while the A1 versus A2 comparison was used to detect the influence of water treatment on resistant rice. In the following analysis, we will compare the P450 family genes and ABC transporter family genes selected in the two groups with those selected in the above four groups, so as to exclude the interference of water solvent.
Functional annotation of DEGs and selected candidate genes
We primarily focused on genes which were differently expressed between treated sensitive and treated resistant plants, which may represent targets for understanding the NTSR mechanism. CYP450 genes have previously been implicated in herbicide metabolism [20], while CYP76 family genes are involved in the metabolism of benzoyl urea herbicides and have been shown to play a detoxification role for benzoyl urea herbicides [8]. Researchers have found that CYP72A31 and CYP81A6 are involved in the detoxification of bensulfuron-methyl herbicides in rice and A. thaliana [9-11]. However, no studies on the role of P450 family genes in the regulation of cyhalofop-butyl resistance have been conducted in L. chinensis using whole transcriptomic methods. In higher plants, ABC transporters have been implicated in detoxification of xenobiotics, including herbicides. Unlike P450, which detoxifies herbicides through metabolic mechanisms, ABC transporters detoxify herbicides and confer herbicide resistance by transporting herbicides and the metabolites that result from their breakdown. The four comparison groups (B1vsA1, B3vsA3, A2vsA3, B2vsB3) which were most likely to be relevant to cyhalofop-butyl resistance were selected for subsequent correlation analyses (Figure 2).
Expression analysis of sensitive versus resistant genotypes without exposure: A1vsB1
A total of 14709 genes were identified in this group, among which 6147 genes were up-regulated and 8562 were down-regulated (Figure 3A). The up-regulated genes were primarily associated with biological processes such as single organism metabolic process, single organism process, and transmembrane transport. These genes were also enriched in molecular functions, such as transmembrane transport activity, transporter activity, and substrate-specific transporter activity and enriched in Cell components such as plasmid and chloroplast (Figure 3B). KEGG pathway analysis showed that some genes associated with metabolic pathways and secondary metabolite biosynthesis pathways were highly expressed in resistant L. chinensis (Figure 3C). Analysis of all induced genes showed that there were 25 ABC transporter family genes with higher expression in the resistant population. Additionally, 39 CYP450 family genes were identified to be more highly expressed in the resistant population. Among them, 32 genes were differentially expressed only in this subset. Two CYP72A family genes (Chr4.g12261 and Chr4.g12260) and two CYP76 family genes (Chr17.g44850 and Chr17.g44847) were identified in this subset, all of which were highly expressed in LC1802. Members of the CYP76 family and CYP72A31 have been identified as herbicide metabolic resistance genes [8-9]. Therefore, we hypothesized that these genes might be impacting the NTSR of LC1802. Additionally, 48 genes related to fatty acid biosynthesis were identified, of which 20 were more highly expressed in the resistant population and 28 were more highly expressed in the sensitive population. Many genes associated with plant hormone signal transduction and photosynthesis were found to be more highly expressed in the resistant population. The DELLA protein has been shown to play a role in the regulation of gibberellin (GA) signaling by suppressing stress response pathways [21]. This gene is also associated with growth inhibition of weeds [22]. Interestingly, three genes encoding DELLA proteins were identified only in this subset of DEGs. Among them, Chr3.g10116 was found to have no expression in the resistant population, despite being present in the sensitive population.
Expression analysis of sensitive vs. resistant genotypes with herbicide exposure: A3vsB3
In this group, 2704 DEGs were identified, among which 2065 genes were up-regulated and 639 genes were down-regulated (Figure 4A). A total of 1426 DEGs only existed in this subset. These genes showed no difference in expression when the two materials were not treated but showed differences in expression during cyhalofop-butyl treatment. Additionally, 1091 genes were differentially expressed in this set as well as the B1 versus A1 comparison, indicating that these genes were differentially expressed under both normal conditions and cyhalofop-butyl treatment. GO term analysis of biological processes resulted in the identification of up-regulated genes related to carbohydrate metabolic process, cell wall organization or biogenesis, and reactive oxygen species metabolism. These genes were also enriched in molecular functions, such as hydrolase activity, hydrolyzing O−glycosyl compounds, hydrolase activity, acting on glycosyl bonds and enriched in Cell components such as cell periphery and extracellular region (Figure 4B). KEGG pathway analysis results were similar to the results seen in the B1 versus A1 comparison, and a total of 180 up-regulated genes were identified as being involved in different metabolic pathways. 130 were involved in secondary metabolite biosynthesis pathways (Figure 4C). These findings further support our hypothesis that metabolic genes and transport genes mediate the high cyhalofop-butyl resistance in the LC18002 line. Additionally, 11 ABC transporter family genes were identified among the DEGs, 9 of which were differentially expressed only in this subset. Among the up-regulated genes, we found that ABCA7 (Chr2.g06140) and ABCB2 (Chr10.g32569) were highly induced and may represent candidates for further study of cyhalofop-butyl resistance in LC18002. CYP450 family genes have been shown to play a crucial role in herbicide metabolism [20], and we found 16 CYP450 family genes which had higher expression levels in LC18002. In particular, we found that a CYP450 gene (Chr8.g25254) and CYP84A1 (Chr10.g30537) were up-regulated not only in this comparison, but also in B1 versus A1, despite showing no differential expression in any other comparisons. Given that other members of the CYP450 family have been implicated in herbicide tolerance, higher expression of these genes may play a role in the resistance of LC18002 to herbicides. Additionally, we found 12 possible xyloglucan endo-transglucosylase/hydrolase encoding genes, and other genes of this class have previously been reported to participate in abiotic stress in pepper [23] and tea [24]. In addition to the above genes, some other key genes involved in herbicide resistance pathways were also found in this subset. Pectate lyases have been shown to play an important role in herbicide resistance by regulating the composition of polysaccharides in plants to affect plant stress tolerance [25]. Two pectate lyase genes were identified in this subset, both of which were highly expressed in LC18002. A protein strubbelig-receptor family 7 gene was also identified, and its homolog has been shown to be a sensor for herbicides, mediating intercellular and intracellular abscisic acid (ABA) pathways to regulate herbicide stress signaling [26]. Two aquaporin nip5-1 genes were also found to be more highly expressed in LC18002, and members of this class are known to control pore size with significant impacts on herbicide tolerance. Peroxidase 12-like genes have also been shown to play a part in abiotic stress tolerance and herbicide resistance by regulating the jasmonic acid biosynthesis pathway [27-28]. Pectinesterase inhibitor 34 is involved in fine-tuning cell wall remodeling processes under abiotic stress [29] and three up-regulated pectinesterase inhibitor genes were identified in this group, two of which were differentially expressed only in this subset.
Effects of herbicide exposure on the sensitive genotype: B3vsB2
A total of 1649 DEGs were identified in this group, among which 658 genes were up-regulated and 991 genes were down-regulated after herbicide treatment (Additional file 2A). This group can be used to investigate which genes were differentially expressed following cyhalofop-butyl treatment in the sensitive population. GO term analysis of biological processes indicated that many up-regulated genes were related to protein phosphorylation, phosphorylation, phosphorus metabolic process, and phosphate containing compound metabolic process. These genes were also enriched in molecular functions, such as small molecule binding, nucleotide binding and nucleoside phosphate binding and enriched in Cell components such as membrane, intrinsic component of membrane and integral component of membrane. (Additional file 2B). KEGG pathway analysis showed that thirteen genes with higher expression after cyhalofop-butyl treatment were involved in plant hormone signal transduction (Additional file 2C), including indole-3-acetic acid amido synthetase, TF PIF5, and regulatory protein NPR5. Seven genes were involved in glycerophospholipid metabolism process, while five genes were involved in fatty acid degradation such as alcohol dehydrogenase and amino acid permease. Five other genes were involved in valine, leucine, and isoleucine degradation pathways. Eight ABC transporter family genes with differential expression were identified in this group. Among them, seven were induced and one was repressed. The induction of these different genes in the sensitive population likely represents a response to stress caused by cyhalofop-butyl treatment, including ABC transporters being used to transport active herbicide molecules.
Effects of herbicide exposure on the resistant genotype: A3vsA2
A total of 150 DEGs were identified in this group, among which 96 genes were up-regulated and 54 genes were down-regulated (Additional file 3A). This group can be used to investigate gene expression changes caused by cyhalofop-butyl treatment in the resistant line LC18002. As shown in Additional file 3B, among the up-regulated genes, twelve genes were annotated as being involved in stress response, nine genes were enriched in defense response, and nine other genes were enriched in response to acid chemical process. These genes were also enriched in cellar components, such as membrane part and intrinsic component of membrane. KEGG analysis showed that four up-regulated genes were annotated as being involved in metabolic pathways and three genes were involved in the biosynthesis of secondary metabolites (Additional file 3C). Out of the 150 genes which were differentially expressed during cyhalofop-butyl treatment, 28 showed expression change after herbicide exposures exclusively in LC18002. Additionally, three CYP450 genes related to xenobiotic metabolism were highly expressed in the herbicide-exposed resistant line. The expression levels of these genes were relatively constant between the two materials prior to treatment but their expression was significantly induced in the resistant line after treatment with cyhalofop-butyl. It therefore is likely that these genes play some roles in the herbicide resistance of LC18002, although further research is necessary to confirm this.
Effects of water exposure on the sensitive genotype: B2vsB1
This group can be used to identify the effects of water treatment on gene expression in sensitive populations. A total of 17283 DEGs were identified in this comparison, among which 7275 genes were up-regulated and 10008 genes were down-regulated 24 hours after treatment with water (Additional file 4A). GO biological process enrichment analysis showed that among the up-regulated genes, 2004 genes were enriched in single organism process and 1198 genes were enriched in single organism metabolic process. Molecular function analysis showed that 355 up-regulated genes were enriched in transmembrane transporter activity, 278 genes were enriched in substrate-specific transmembrane transporter activity, and 145 genes were enriched in phosphoric ester hydrolase activity (Additional file 4B). Since GO enrichment analysis showed that many up-regulated genes had transmembrane transport activity, ABC transporter family genes were screened in this subset. A total of 29 ABC transporter family genes were found to be up-regulated 24 hours after water treatment, 27 of which were only present in this comparison. Although both water treatment and cyhalofop-butyl treatment affected the expression of ABC transporters, the actual genes affected differed. This result implies that the water present in the cyhalofop-butyl solution is not the sole reason behind the induction of ABC transporters seen under its application.
Effects of water exposure on the resistant genotype: A2vsA1
This comparison group can be used to identify the effects of water treatment on gene expression in LC18002. A total of 749 DEGs were identified in this comparison, among which 137 genes were up-regulated and 612 genes were down-regulated 24 hours after treatment with water (Additional file 5A). Among the up-regulated genes, 7 genes were annotated as participating in the protein folding process, 3 genes were associated with response to light intensity, and 3 other genes were implicated in response to heat. Cellular component analysis indicated that 51 genes were associated with intracellular part, 45 genes were associated with membrane bounded organelle, 45 genes were associated with intracellular membrane bounded or ganelle and 23 genes were associated with the plastid (Additional file 5B). Unlike the A2 versus A3 comparison, only 3 CYP450 family genes were identified in up-regulated genes of this subset which had no differential expression in group A2 versus A3, indicating that the type of CYP family genes involved in water application and cyhalofop-butyl metabolism was different. In addition, an ABC transporter family gene was found to be up-regulated 24 hours after water treatment but was not found to be differentially expressed in the A2 versus A3 comparison. These results again indicate that water treatment alone results in a significantly different response compared to cyhalofop-butyl treatment.
Phylogenetic analysis of candidate genes
Phylogenetic analysis was performed to determine whether the candidate genes identified had close homologs in other species or belonged to any known gene families. The protein sequences of all differentially expressed P450 family genes were compared against P450 genes from A. thaliana, rice, soybean, sorghum, and maize. This clustering analysis revealed that L. chinensis P450 genes clustered closely with P450 genes of other species (Figure 5). Chr8.g25254 was found to cluster closely with Cytochrome P450 71T4-like genes. Chr16.g43989 was annotated as a member of the CYP734A6 subfamily, but phylogenetic analysis shows that it is clustered closely with the CYP72A subfamily of rice. Additionally, we also included herbicide metabolism genes previously studied, such as OsCYP81A6 and OsCYP72A31P. This analysis revealed five genes that were closely related to OsCYP81A6, and seven genes that were closely related to OsCYP72A31P. In addition, we selected one DELLA protein and two pectate lyase proteins as the outgroup and found that they produced two independent branches.
qRT-PCR validation of RNA-seq expression patterns
In order to confirm the magnitude of differential gene expression obtained by transcriptomic approach, the expression levels of 8 genes were measured in two L. chinensis accessions using quantitative real-time PCR (qRT-PCR) (Figure 6). The primers utilized to amplify candidate genes were designed based on the sequence of the identified genes and are listed in Additional file 8. There is a high level of correlation was found between the RNA-seq and qRT-PCR data.
{kind=link}
{kind=link}