Barley plants display low-Pi symptoms at the morphological and molecular levels
Severe low-Pi responses were induced in the barley plant line Rolap grown in the soil containing 8 mg P/kg. P undernourishment caused over 2-fold reduction of plant shoot biomass (Fig. 1A). Shoot fresh weight of plants at 23rd day post-sowing (dps) was significantly reduced, in comparison with control plants, with average mass 8.8 g for stressed plants and 18.5 g for plants growing under Pi-sufficient conditions (p = 0.001) (Fig. 1B). We observed a significantly decreased concentration of Pi ions, with only 0.48 µmol Pi per g of fresh root weight (FW) and 4.2 µmol Pi per g of shoot FW, when compared with the control plants having 3.84 (p = 0.0056) and 24.35 µmol Pi/g FW (p = 0.0001), respectively (Fig. 1C). To examine the induction of changes at a molecular level by low-Pi stress in barley plants, we measured the absolute gene expression of the low-Pi-responsive marker gene IPS1. The barley IPS1 gene is highly expressed under Pi-deficient conditions in the plant line Rolap. At the tillering stage (23 dps), we detected 4191 copies of IPS1 RNA for low-Pi treated roots, normalized per 1000 copies of ADP-RIBOSYLATION FACTOR 1-LIKE (ARF1) reference gene, in comparison to the control plants, with only 58 copies of IPS1 RNA (p = 0.00006) (Additional file 1). Taking validated plant material, we performed tripartite deep-sequencing analysis to: (i) identify Pi-responsive sRNAs, (ii) elucidate changes in the barley transcriptome upon Pi starvation, and (iii) identify mRNA targets for Pi-responsive sRNAs through degradome sequencing (Fig. 2).
Barley plants express an organ-specific set of microRNAs in response to low-Pi conditions
We performed small RNA deep-sequencing to find out which small RNAs are up- or down-regulated by Pi starvation in barley shoots and roots. The average of 30.4 mln reads for roots and 25.2 mln reads for shoots were generated in 50 nt single-read Illumina sequencing (Additional file 2). After adapter and quality trimming, we mapped reads to the miRBase Sequence Database (release 22) to annotate miRNA-derived sequences [63]. A set of parameters were used to define the pool of differentially expressed miRNAs: (i) no mismatches with the reference sequences in the miRBase were allowed; (ii) different types of miRNA sequences were permitted, whether they were annotated as precursor, mature, or isomiR; (iii) miRNA sequences were named accordingly to the name of the assigned reference miRNA; and (iv) significance of fold change (p-value < 0.05) was additionally verified using a restricted Bonferroni p-value test (Fig. 2).
We found 138 and 162 differentially expressed miRNAs (DEMs) annotated to the miRBase (p-value < 0.05) in barley roots and shoots, respectively. Only 25 DEMs were expressed in both examined barley organs (Additional file 3). However, restricted Bonferroni p-value correction narrowed down set of miRNAs to 15 in shoots and 13 in roots (Fig. 3A, left panel). We focused further only on those that passed the Bonferroni test (Fig. 3A, right panel). In both organs, most of the DEMs were significantly up-regulated. Interestingly, out of 15 miRNA, only miRNA166d was down-regulated in shoot under low-Pi (log2(fold change) = -1.18). In our previous work, we showed that miRNA166 is expressed in barley during different developmental stages reaching the highest level in 2-week-old plants [64]. miRNA166 plays an important role in plant development, including root and leaf patterning, by targeting mRNA encoding HOMEODOMAIN LEUCINE-ZIPPER CLASS III (HD-ZIP III) transcription factors [65]. Similarly, only miRNA319b out of 13 DEMs was down-regulated in low-Pi treated roots (log2(fold change) =-1.28). In a previous study, we presented data that Arabidopsis miR319 is a multi-stress responsiveness miRNA [22]. For example, MIR319b gene expression was down-regulated in response to drought, heat, and salinity, but up-regulated in response to copper and sulfur deficiency stresses [22].
Uncharacterized miRNAs are highly induced by low-Pi stress in barley roots
A specific set of miRNAs was expressed in barley shoot or root under low-Pi (Additional file 3). In shoot, only two miRNA families, miRNA399 and miRNA827, were induced, while in root we observed a more diverse response (Fig. 3A, right panel). Apart from miRNA399/miRNA827 induction, we found the following additional miRNA to be up-regulated in root: two miRNA5083 (id = 3, and id = 4), miRNA1511 (id = 6), two miRNA9779 (id = 16, and id = 17), two miRNA156 (id = 65, and id = 69), and miRNA5072 (id = 118). Among these eight miRNAs, only miR156 has been reported before as Pi-responsive in Arabidopsis [42, 55]. The miR156 isomiRs were also found dysregulated in shoot, but none of them pass the Bonferroni test. Our results suggest that there is a more complex response to low-Pi stress regarding miRNA expression in roots than in shoots, where the miRNA action is directed to control the transcript level of either PHO2, SPX-MFS1, or SPX-MFS2 by just two miRNA families.
In order to determine the potential cleavage activity of miRNAs identified in root we performed degradome analysis. None of these eight miRNAs were found to recognize a specific target from the pool of barley mRNAs. Our result raises the hypothesis that plants may release Pi from the pool of stress-responsive small RNAs. In addition, most of them match to the pre-miRNAs, not mature molecules (Fig. 3A, right panel).
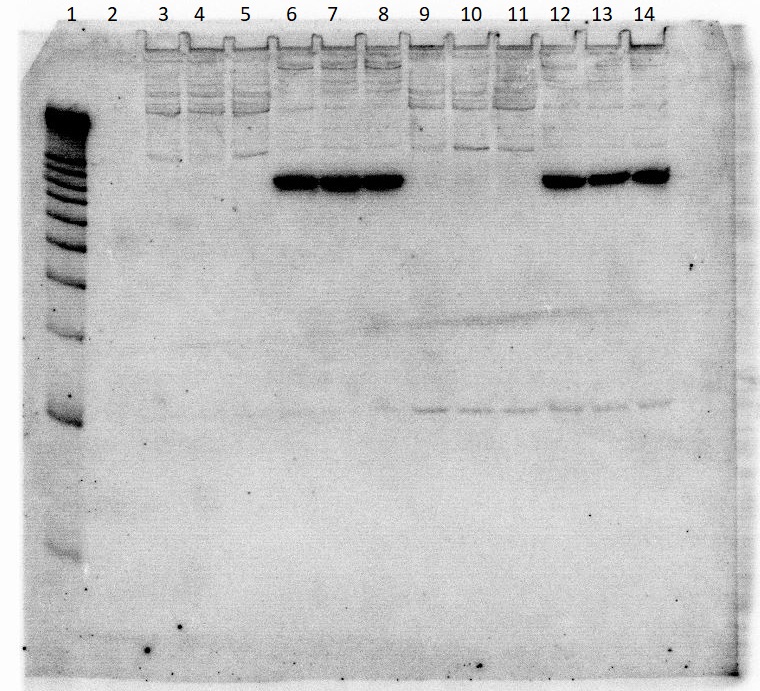
NGS (Next Generation Sequencing) data were validated by complex analysis of mature miR827 (from 3’ arm) in all samples taken for deep sequencing. The absolute expression level of miR827 is significantly up-regulated in both shoots and roots under a low-Pi regime (Fig. 3B). The log2 fold change of miR827 molecules defined by deep-sequencing in shoot (id: 2073) was found on the same level in root (id: 114), log2(fc) = 3.05 and 3.01, respectively (Fig. 3A). The ddPCR results were consistent with NGS data showing up-regulation of mature miR827 molecule in both tested organs (Fig. 3B). These data were confirmed by northern blot hybridization (Fig. 3C) and degradome analysis (Fig. 3D). The results showed that SPX-MFS1 transcripts have been recognized and cleaved by Ago protein associated with miR827 in barley (p = 0.014) (Fig. 3D).
Different classes of small RNAs in barley accumulate in an organ-specific manner under low-Pi regime
The small RNA reads which did not map to miRBase were mapped to particular classes of barley cDNAs derived from the Ensembl Plants database (release 40), both separately and to all of them (Fig. 2). All sequences mapped to barley cDNAs are listed in Additional file 3. We found that small RNAs, other than miRs, differentially expressed (DESs) in barley under Pi starvation were represented by 199 unique sequences (0.01 % of all unique reads from barley shoots (Additional file 4) and by 1796 (0.13 %) unique reads from roots (Fig. 4A, Additional file 5).
We analyzed whether different lengths (taking sequences from 18 to 25 nt in lenght) and classes of small RNAs contributed to either root or shoot response to low-Pi conditions. In roots, the length distribution of DESs remained balanced, from 10.91 % for the representation of 24 nt sequences to 15.26 % for the 18 nt sequences, which were the most abundant (including 274 DESs) (Fig. 4B). In shoots, the representations of DES lengths fluctuated more than in roots. The 19 nt sequences were the most visible (21.11 %), while three representations did not score more than 10%: the 22 nt (9.55 %), 23 nt (8.54 %), and 25 nt (3.52 %) sequences (Fig. 4B, Additional file 6).
In roots, 1070 unique reads were mapped to cDNA sequences annotated in the Ensembl Plants databases (non-translating, protein-coding, pseudogenes, rRNA, snoRNA, snRNA, sRP-RNA, tRNA), while 726 unique sequences remained without match. The DESs obtained from low-Pi roots were mostly annotated to protein-coding mRNAs (38.54 %), rRNAs (34.17 %), and non-translating RNAs (19.49 %). Below 5 % of overall DESs, we found a number of remaining cDNA classes, such as snoRNAs (2.49 %), tRNAs (2.47 %), SRP-RNAs (1.17 %), snRNAs (0.95 %), and pseudogenes (0.65 %). While in shoot, we found 199 DESs under the low-Pi regime. Altogether, 116 out of 199 differentially expressed small RNAs (DESs) were annotated to the barley Ensembl Plants database, where 83 sequences remained without match (Additional file 4). In the case of shoot samples, 85 % of annotated DESs represented only protein-coding mRNAs (47.87 %) and non-translating RNAs (36.49 %) (Fig. 4A, right panel; Additional file 7). We did not find any DESs annotated to the snRNAs, SRP-RNAs, or tRNAs from barley shoot upon low-Pi. In addition, total numbers of 166 DESs (83 %) in shoots and 1560 DESs (87 %) in roots were significantly up-regulated after exposure to low-Pi stress (Additional file 4, Additional file 5, Fig. 7).
Among the unannotated reads of sRNAs in roots, the highest fold change was observed for a 19 nt 5’-ACCTACTCGACCTCGGCCG-3’ molecule (log2(fold change) = 8.02, induction) and a 22 nt 5’-CTAATACCGGATACGCGAACCG-3’ molecule (-5.87, repression). The BLAST (Basic Local Alignment Search Tool) analysis of first (19 nt) molecule showed a perfect match to either the intergenic region of barley chromosome no. 5, soil bacteria (mesorhizobium), or Linum usitatissimum L., while the second molecule (22 nt) mapped to RNA encodes 16S rRNA. Furthermore, in roots, the most abundant small RNA was a 25 nt 5’-ACCGACCTACTCGACCCTTCGGCCG-3’ molecule (15847.7 and 65590.5 mean of normalized counts in barley root in control and low-Pi conditions, log2(fc) = 2.82). This small RNA matched several barley loci encoding SSU (small subunit) rRNAs (Additional file 5).
In our results from low-Pi treated shoot samples, the highest fold change was represented by a 24 nt 5’-AAGATTGGTTGGTTGGTTGGGTCT-3’ molecule (log2(fc) = 8.72, induction). This 24 nt molecule is a part of transcript encoding a putative pentatricopeptide repeat (PPR) protein. The PPR protein family facilitates the processing, splicing, editing, stability, and translation of RNAs in plants [66]. The most abundant small RNA was a 19 nt 5’-GGGCCTGTAGCTCAGAGGA-3’ molecule (9471.5 and 49914.1 normalized mean counts in barley shoot in control and low-Pi, respectively, log2(fc) = 2,45). This sRNA was mapped to the barley genomic loci (EPlHVUG00000039813), which encodes arginyl-tRNA (trnR-ACG) and a cDNA encoding uncharacterized protein (HORVU2Hr1G084630) which is likely involved in carbon fixation. Interestingly, the pool of DESs was selective, considering organ-specific expression change, providing only three unique sequences that were significantly changed in both barley organs under low-Pi regime (Fig. 4A, left panel). These molecules were: (i) 20 nt 5’-AGTAGAGGTCGCGAGAGAGC-3’ (log2(fc) = 2.01 in root and 1.16 in shoot, respectively) annotated to the 26S rRNAs, (ii) 24 nt 5’-ATTCTCCGCGTCGGATACCTGAGA-3’ (3.69 in root and 2.07 in shoot) encoding the barley MYB21 transcription factor, and (iii) 21 nt 5’-TGCCAAAGGAGAACTGCCCTG-3’ (4.64 in root and 6.27 in shoot) mapped to the intergenic region of barley chromosome no. 3 (Additional file 4).
The results obtained in this study show again that barley roots exhibit a more diverse pool of Pi-responsive small RNAs which may trigger developmental adaptation of the root to Pi-starvation. Additionally, 613 rRNA-derived sRNAs are up-regulated, whereas 176 rRNA-derived sRNAs are down-regulated in barley roots (Additional file 5). We believe that such sRNA may be further processed, serving as a Pi source to compensate Pi deficiency.
Identification of barley genes responsive to Pi-starvation
Since we observed, that most of the other sRNAs in shoot were derived from either protein-coding mRNAs or non-translating RNAs, we checked whether this observation is correlated with gene expression changes of polyadenylated RNAs in barley shoot under Pi-starvation. Among 98 of identified DEGs, the transcripts of 56 annotated loci were significantly up-regulated, while those derived from 42 loci were down-regulated in Pi-starved barley shoots (Additional file 8). Repressed loci were found to be preferentially located at barley chromosome no. 2, while induced loci were found mostly at barley chromosomes no. 3 and 5 (Fig. 5B).
The highest enrichment of DEGs was found in the GO terms, either (i) belonging to the cellular components of the chloroplasts; (ii) showing catalytic activity, either ion or metal binding properties; and (iii) involved in the various biological and metabolic processes related to stress response and plant defense (Fig. 5A). A major set of up-regulated DEGs represented genes involved in the Pi signaling. Among them, we found genes encoding: IPS1 (log2(fc) = 5.89) [54], inorganic pyrophosphatase (PPase, 4.01) [67], SPX-domain containing protein 5 (SPX5, 3.44) [68], phosphate transporter PHOSPHATE 1-3 (PHO1-3, 2.97) [69], SPX-MFS2 (2.79) [56], haloacid dehalogenase-like hydrolase (HAD1, 1.95), [70] and five different purple acid phosphatases (PAPs) (Fig. 5C; Additional file 8) [71]. Interestingly, four genes were induced to a higher extent than the low-Pi stress marker, IPS1 gene. These genes encode ferredoxin (FD1, log2(fc) = 14.20), mitochondrial-processing peptidase (13.35), chlorophyll a/b binding protein (8.90), and alpha-amylase (7.30), and are engaged in photosynthesis, redox reactions, reactive oxygen species (ROS) homeostasis, and co-ordinated mobilization of nutrients. Chloroplasts and mitochondria are the organelles with the highest Pi requirements. Strong FD1 gene up-regulation most likely reflects the accumulation of reduced ferredoxin in chloroplasts. Low-Pi lowers the capacity to process incoming light and enhances starch accumulation in chloroplasts, thereby leading to photoinhibition [72, 73]. Within the category of genes that were significantly down-regulated, most of them were related to stress and defense responses (Additional file 8); for instance, uncharacterized protein (HORVU2Hr1G030090, -6.50), oxalate oxidase (-4.41) [74], beta-sesquiphellandrene synthase (-3.41), glutamate carboxypeptidase (-3.17), chalcone synthase (-3.05) [75], or caleosin-like protein (-2.95) (Fig. 5C). Only two repressed genes are known to be directly involved in Pi signaling and metabolism, SPX-MFS1 (-2.58), targeted by miR827 [57] and probable inactive purple acid phosphatase (-1.75). Additionally, two genes encoding laccases (LAC19-like, Additional file 8), cell wall-localized multi-copper oxidases, were significantly down-regulated (-2.10 and -2.44) in our mRNA RNA-Seq data. Laccases are involved in copper homeostasis (Fig. 7) and lignin biosynthesis, and have been shown to be targeted by miR397 in maize [76] and Arabidopsis [77]. Furthermore, key genes encoding proteins involved in the nitrate and phosphate cross-talk were affected by low-Pi conditions in barley shoots, such as NIGT1 (NITRATE-INDUCIBLE, GARP-TYPE TRANSCRIPTIONAL REPRESSOR 1) transcription factor (3.80) [78, 79] and nitrite reductase (1.98), as well as high-affinity nitrate transporter NRT2.1 (NITRATE TRANSPORTER 2.1) (-2.60) [80].
Absolute quantification of a few selected transcripts was performed to validate RNA-Seq data obtained in this study. Two genes which were highly induced (encoding endonuclease S1/P1 and 3’-5’-exonuclease) and two which were severely repressed (encoding oxalate oxidases) under the low-Pi regime were taken for ddPCR (droplet digital PCR) analysis (Fig. 6A). We confirmed statistically significant changes (p < 0.05) in normalized copy number (per 1000 copies of the ARF1 reference gene) of all genes taken for analysis.
Pi-responsive motifs found in the promoters of DEGs
In general, genes that are affected by Pi status possess characteristic cis-regulatory elements within either promoter or 5’-UTR regions [81]. Previously, we have shown the importance of the P1BS motif (PHR1 binding sequence, consensus GnATATnC, [82]) and P-responsive PHO elements (consensus ATGCCAT, [83]) in the expression efficiency of the barley PHO2 gene [48]. Both motifs may bind PHR-like (PHOSPHATE STARVATION RESPONSE) transcription factors (TFs) and act as activators or repressors of downstream gene expression in a Pi-dependent manner [84]. Likewise, we hypothesized that regulatory regions of the identified DEGs had Pi-responsive motifs, which may be bound by PHR TFs, causing gene expression dysregulation. To confirm this hypothesis, we analyzed DNA sequences from the 2000 bp region upstream of the predicted transcription start sites from all 98 DEGs (Additional file 9). In the next step, promoter data were directly screened for P1BS and P-responsive PHO element consensus sequences by multiple promoter analysis using the PlantPAN3.0 tool [85]. We confirmed the presence of Pi-dependent motif in 55 out of 98 DEGs promoters. An in silico approach detected 46 DEGs having at least one P1BS motif (Additional file 10) and 17 DEGs with at least one P-responsive PHO element (Fig. 6B, Additional file 11). The most over-represented motifs were found in the promoters of genes encoding sulfoquinovosyl transferase SQD2-like (log2(fc) = 3.74) [86], phosphoenolpyruvate carboxylase 1-like (log2(fc) = 1.73) [87], and pyridoxal phosphate-dependent transferase (log2(fc) = -2.61) [88]. Each of the genes harbor three P1BSs and one P-responsive PHO element, as well (Additional file 8).
Degradome profiling describes post-transcriptional regulatory network of identified DEMs
After identification of (i) differentially expressed miRNAs (DEMs), (ii) other sRNAs (DESs), and (iii) mRNAs (DEGs), we used this comprehensive data together with cDNAs annotated in the Ensembl Plants database to identify the sRNAs directly involved in RNA degradation. The DESs were also examined, because we assumed that there may have been putative miRNAs that were not mapped to the miRbase, due to restricted query settings allowing no mismatch. Molecules which exhibited a single mismatch (or more) may still function as miRNA in barley. Degradome libraries were carried out for root, as well as for shoot, and sequenced using an Illumina System. The received data were analyzed using two independent in silico approaches (Fig. 2). At times, the different algorithms used elicited different miRNA targets; however, the general degradome pattern was equivalent for both approaches (Fig. 3A, right panel, Additional files 12-24).
First, we searched for the potential target mRNAs for a broader set of differentially expressed miRNAs with significant fold change (p-value without Bonferroni correction), taking 162 DEMs from shoot and 138 DEMs from root, respectively (Additional file 3). A total of 1964 scores were obtained for shoot DEMs (1303 using the TargetSeek approach and 661 using PAREsnip2) (Additional file 12, Additional file 16), while in root there were 1515 records (921 and 594, respectively) (Additional file 14, Additional file 20, Additional file 21). Accordingly, shoot examination proved the proper selection of miRNAs from all differentially expressed small RNAs, as all 661 records predicted by the PAREsnip2 approach were categorized with the best score (0). A majority of records corresponded to different miR399 and/or miR827 isomiRs and their known targets PHO2 or SPX-MFS1/SPX-MFS2, respectively. In shoot, the best scoring miRNA:mRNA match was found for mature miRNA399d (21 nt, id = 2066), which guides cleavage within the 5’-UTR of PHO2 mRNA (isoform no. 3) in position 857 (TargetSeek: score = 0, MFE = -41,1, PAREsnip2: score= 0, MFE = -41.1) (Table 1). The miR5049e (21 nt) targets various isoforms of mRNA encoding ENT domain-containing proteins with high probability (TargetSeek: score = 1.5, MFE = -27.4), which may be involved in the plant defense response to fungal infection and anthocyanin biosynthesis [89, 90]. Moreover, we found two target mRNAs (HORVU3Hr1G094730, HORVU6Hr1G031450) encoding SPL-like TFs guided for cleavage by two miR156 isomiRs (id: 2053 and 2054) in barley shoot (Additional file 16, Additional file 17). The SPL genes are post-transcriptionally regulated by miR156 and control shoot branching in rice [91].
In contrast, in roots, where the pool of identified DEMs is more diverse, the PTGS regulatory network involves many TFs with various functional domains (Additional file 14). For instance, the 21 nt mature miR444b (id = 101) targets mRNA encoding MADS-box transcription factor 57 (HORVU6Hr1G073040, TargetSeek: score = 0, MFE = -40.1, PAREsnip2: score = 0, MFE = -40.1), which regulates long-distance nitrate transport and root elongation in rice [92] (Table 1). The well-known abiotic stress-responsive miR319c (21 nt) [6] was found to target various isoforms of GAMyb TF (HORVU3Hr1G079490, TargetSeek: score = 5, MFE = -34.6, PAREsnip2: score = 3, MFE = -35.4). The same predicted miR319 target has been previously found in RNA-Seq of Pi-starved tobacco plants [93]. Its targets, the plant GAMyb TFs, have been shown to activate gibberellin-responsive gene expression of α-amylase in barley [94, 95]. Our degradome data indicate the possible cleavage of various mRNA isoforms encoding nuclear transcription factor Y subunit A-4 (NFYA4, HORVU4Hr1G005670, TargetSeek: score = 3, MFE = -27.6, PAREsnip2: score = 2, MFE = -28.1) mediated by miR169d (20 nt, id = 84) (Table 1), what is consistent with previous studies on Cunninghamia lanceolata L. showing that NFYA4 mRNA is targeted by miR169d (authors used PAREsnip approach) [96]. The NFYA4 protein is involved in many abiotic stress responses and may regulate the timing of transition from vegetative to reproductive phase [97]. All predicted DEM targets for both organs are listed in Additional files 13, 15, 17, and 21.
Putative regulatory small RNAs identified in degradome data
Degradome profiling was performed to test whether any of the sequences from the 1796 DESs found in roots or 199 DESs found in shoots contribute to the complexity of gene regulation during low-Pi stress. A total of 759 records (245 using the TargetSeek approach and 514 using PAREsnip2) were found in the degradome profiles matching root DESs (Additional file 15, Additional file 22, Additional file 23) and 160 records (87 and 73, respectively) matching shoot DESs (Additional file 13, Additional file 18, Additional file 19). Taking only either the most up-regulated or the most down-regulated sRNAs for degradome screening, we found six promising target genes in shoot and five in root (Table 2). For example, in roots, the highly up-regulated 20 nt sequence 5’-ACCGACCTACTTGACCCTTC-3’ (log2(fc) = 6.46, id = 348) binds to the 3’-UTR region of the MYB44 TF’s mRNA and guides/promotes cleavage in the 1037 position (PAREsnip2: score = 4; MFE = -33.3) (Table 2). RNA-Seq data for potato (Solanum tuberosum L.) proved that expression of the MYB44 gene is highly downregulated under low-Pi in roots [98], which may be the result of miRNA-guided PTGS. Studies in potato have indicated that MYB44 TF may form a regulatory complex together with WRKY6 TF, which negatively regulates Pi transport by suppressing PHO1 expression [98]. Other degradome records, among the most differentially expressed sRNAs, were found to target mRNAs of the V-ATPase assembly factor (VMA21-like) and three barley genomic loci encoding uncharacterized proteins (HORVU7Hr1G053570, HORVU1Hr1G027340, and HORVU0Hr1G023910) (Table 2). For example, the potential cleavage activity was predicted for 24 nt sequence 5’- AGAGGAAACTCTGGTGGAGGCTCG-3’ (log2(fc) = -3.58, id = 463), which may target the mRNA encoding uncharacterized protein with unknown PTHR47188 domain (Fig. 4C).
Analogous degradome screening was done for shoot data. Among all identified DESs, we found that the most upregulated sequence, 24 nt 5’- AAGATTGGTTGGTTGGTTGGGTCT-3’ (long2(fc) = 8.72, id = 2112), targets the 3’-UTR of mRNA encoding multiple organellar RNA editing factor 9 (MORF9, HORVU7Hr1G073170, TargetSeek: score = 16.5, MFE = -28.1) (Table 2). MORF9 proteins are required for RNA editing in plastid mRNAs, which may contribute to stress adaptation in plants [99, 100]. In both approaches, we found that the 21 nt sequence 5'-TGCCAAAGGAGAACTGCCCTG-3’ (6.27, id = 2265) targeted the same isoform of PHO2 mRNA (HORVU1Hr1G085570.3, TargetSeek: score = 6, MFE = -29.9, PAREsnip2: score = 3.5, MFE = -33.4). When we browsed the miRBase using this 21 nt sRNA as a query, we found high similarity to the osa-miR399a, exhibiting only one mismatch. Thus, we suspect that such sRNA may function as another miR399 isomiR in barley. Most dysregulated DESs were also found to target mRNAs encoding methyltransferase type 11 domain-containing protein (MT11), AAA-ATPase (At3g50940-like), lysine-specific demethylase 5A (LSD), or an uncharacterized protein with a predicted transmembrane domain (HORVU3Hr1G036970). The best scoring records were found for the 21 nt sequence 5’- TTAGATGACCATCAGCAAACT-3’ (3.11, id = 2279), which targets mRNAs encoding SPX-MFS1 (PAREsnip2: score = 2.5, MFE = -27.5) and SPX-MFS2 (TargetSeek: score = 3.5, MFE = -26) (Table 3). This result suggests that such sRNA may exist as another miR827 isomiR. Moreover, this is consistent with the screening made for differentially expressed miRNAs, where miR827 targeted both SPX-MFS proteins, depending on the approach we used (Fig. 3A). In addition, the 18 nt sequence 5’- AATCGTCTTTACATCGGATG-3’ (3.24, id = 2117) was found to target mRNA encoding glutaminyl-peptide cyclotransferase (HORVU7Hr1G003920, PAREsnip2: score = 2.0, MFE = -19.6) (Fig. 4C), which may be involved in plant defense reactions [101].
Some other interesting Pi-related targets which are recognized by DESs were found in our root degradome data, but the prediction scores were weaker than those in the examples described above. For instance: nitrate reductase (HORVU6Hr1G003300), high-affinity nitrate transporter-activating protein (HORVU5Hr1G115500), MYB-like TF (HORVU7Hr1G027370), and stress-induced TF NAC1 (HORVU5Hr1G111590) were found. Interestingly, among the 98 DEGs identified in this study, only two of them (SPX-MFS1 and SPX-MFS2) were found as putative targets of miRNA guided activity. In addition, none of the DEGs gene IDs were found to match with any of the identified IDs classified for differentially expressed small RNAs. All predicted DESs targets are listed in Additional files 14, 16, 19, and 23.
{kind=link}
{kind=link}