Inheritance of dwarfing traits
Phenotype of dwarfing traits, tree height, trunk diameter, and canopy width segregated significantly among ‘Red Fuji’(Malus × domestica Borkh.)scions grafted on the 1123 hybrids of ‘Malus baccata’ × ‘M9’ (Fig.1 A and B; Table 1). None of the frequency distributions of these dwarfing traits showed a Gaussian distribution (Fig. 2 A-C). Broad sense heritability was estimated to be 73.0%, 79.7%, and 82.3% for tree height, trunk diameter, and canopy width, respectively (Table 1).
Significant linear regressions were shown between tree height, diameter and canopy width. The Pearson’s correlation coefficients were 0.9421, 0.8663, and 0.9388 (P<0.01) for tree height/trunk diameter, tree height/canopy width, and trunk diameter/canopy width, respectively (Fig. 2 D-F).
Genetic of ultra affinity and extremely dwarfing group selection
The major segregation give the present of metaphase trend in those three dwarfing phenotype, but there still a minor group cover the range from 4.5% to 5.4% holding an ultra affinity in genetic(Fig. 2 A-C), which will help the selection of extremely dwarfing group both in validation of dwarfing markers and new strain of dwarfing rootstock. An intersection used to cross three dwarfing phenotype, 41 and 44 of individuals were selected between plant height cross trunk diameter and canopy width cross trunk diameter, lastly we get 34 of individuals gathering three dwarfing phenotype in dwarfing group(Fig. 3 A,B,E), and 36 of individuals in vigorous group(Fig. 3 C,D,F). By each group, the top of 30 were selected as extremely dwarfing/vigorous (Fig. 3 B,D,G)respectively to validate the molecular markers and genetic linkage analysis.
Markers validation and genetic linkage analysis
Three SSR markers, Hi01c04, Hi04a08, and CH03a09 near Dw1 on chromosome 5, and the other two markers, Hi07d11 and CH02d08 close to Dw2 on chromosome11, were genotyped in 30 hybrids where the scion exhibited the dwarfing phenotype [2]. All five of these markers were heterozygous in ‘M9’ rootstock, homozygous in ‘Malus baccata’ rootstock, and displayed a distorted segregation ratio in dwarfing F1 hybrids,indicating the association of these markers with dwarfing effects (Fig. 4).
Ratio of partial segregation will give more information between the genetic marker and the functional candidate dwarfing genes. There were explanation of dwarfing phenotype in terms of 86.7%, 76.7% and 73.3%, means the recombination of 13.3%, 23.3% and 26.7% addressed in Hi01c04, Hi04a08, and CH03a09 respectively in Dw1 section(Fig.5 A,C), and the genetic distance were of 12.9cM, 25.2cM and 29.8cM convert into physical distance were 0.15Mb, 0.29Mb and 0.35Mb which were used to cut from the position of SSR marker for compressing. For example, Hi01c04 at the top of the QTL section with the physical position of 4.58Mb, exploration of 86.7%, recombination of 13.3%, genetic distance of 12.9cM, and physical distance of 0.15Mb. Position of 4.58Mb(location of Hi01c04) add up with 0.15Mb is the new top of the section(Fig.5 A,C); Hi04a08 is in the middle of the QTL section with the physical position of 5.15Mb, exploration of 76.7%, recombination of 23.3%, genetic distance of 25.2cM, and physical distance of 0.29Mb, afterwards 5.15Mb cut off with 0.29Mb is the new bottom of the section, then a new small sized section from 4.73Mb to 4.86Mb was cut out from Dw1(Fig.5 A,C). As state above, the intervals were narrowed down based in this protocol, the next section cut out from Dw1 was from 5.44Mb to 7.27Mb, and Dw2 section was compressed from 2.35Mb to 8.63Mb(Fig.5 B). Size of intervals of Dw1 and Dw2 were narrowed down which will assist the prediction of candidate dwarfing genes next step.
Re-sequencing analysis and candidate gene prediction
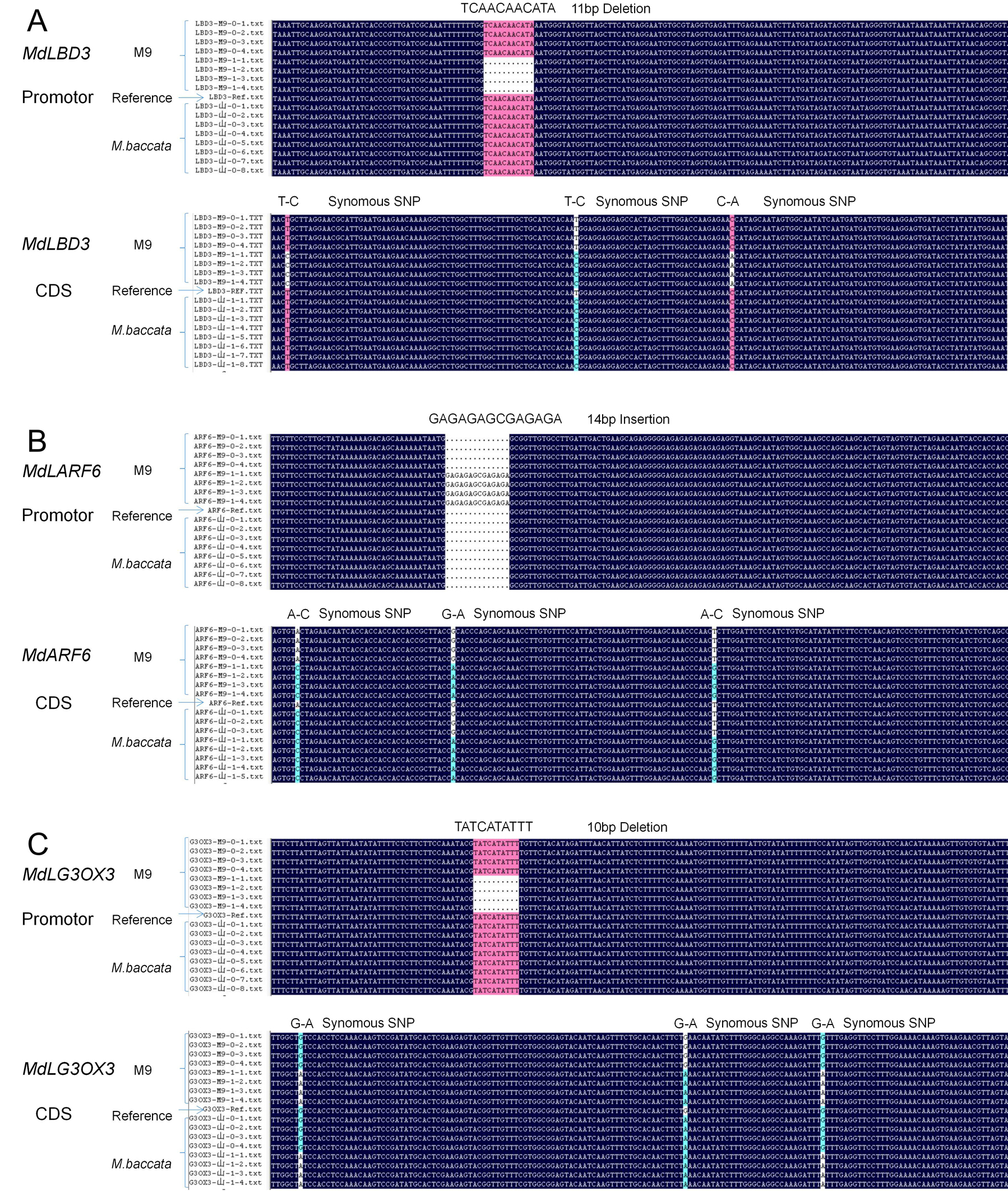
There were a mount of variation in sequence level in terms of SNPs, InDels and SVs, and the mutants were 4103 and 7507 in interval of Dw1 and Dw2 respectively(Table 2), which give a lot of information which troubled in validate sites selection related with dwarfing trait, fortunately the pipe line mentioned in method fulfill the duty to assist validate selection as follows: There were 1350 and 2135 of mutant in narrowed down section of Dw1and Dw2 respectively(Table 2); then 216 and 407 of mutant after constraint by M9(0/1) & Malus baccata(0/0); in turn 43 and 85 of mutant constraint by B9(0/1) & Malus robusta(0/0); 16 and 30 of mutant after confined in promoter and exonic; lastly, four and nine InDel mutant respectively in Dw1 and Dw2 were selected after excluded Synonymous(Table 2,), and markers explored on the InDel variation were also genotyped in the 60 hybrids(Fig.6), the scion grafted on each of the 30 hybrids showed extremely dwarfing or extremely vigorous tree architecture. Whenever a marker recombined with the phenotypes, it’s considered not to be a diagnostic marker at all and the corresponding gene should not be the candidate gene. Of these markers, the segregation ratio of C10 and B06 near Dw1 and M90 close to Dw2 was 30:0 in dwarfing hybrids and 0:30 in vigorous hybrids (Fig. 6). According to the apple genome, C10, B06, and M90 were located in the upstream regions of MdLBD3, MdARF6, and MdG3OX3, respectively. Therefore, these three genes were selected as potential candidate genes for further validation.
Validation of allelic variation of the potential candidate genes
The relative expression of MdLBD3 and MdG3OX3 was significantly higher in the fine roots of ‘M9’ than in ‘Malus baccata’, but MdARF6 was expressed at higher levels in the roots of ‘Malus baccata’ than in ‘M9’ (Figure 7A). The 0~1104 bp, 0~1289 bp, and 0~1176 bp segments upstream of the transcription start site (TSS) of MdLBD3, MdARF6, and MdG3OX3, respectively, were cloned from the DNA of ‘M9’ and ‘Malus baccata’ and Sanger sequenced (Fig. S1; Fig. 7C). Besides some SNPs that were heterozygous on ‘M9’, an 11 bp and a 10 bp deletion were found -339 bp and -278 bp upstream of the TSS of MdLBD3 and MdG3OX3, respectively, while a 14 bp insertion was located -954 bp prior to the TSS of MdARF6 (Fig. 7C). Non-synonymous variations were not found on the coding sequences of these three genes (Fig. S1). To test whether the gene expression differs between genotypes of these InDels in MdLBD3, MdARF6, and MdG3OX3, fine roots of 108 F1 randomly chosen F1 hybrids of ‘Malus baccata’ × ‘M9’ were sampled for a gene relative expression assay (Table 3). The relative gene expression of hybrids with MdLBD3 Del:Ins, MdG3OX3 Del:Ins, and MdARF6 Del:Del genotypes was significantly greater than that of hybrids with MdLBD3 Ins:Ins, MdG3OX3 Ins:Ins, and MdARF6 Del:Ins genotypes, respectively (Fig. 7B). These promoter variants of each gene were then constructed fusion with a GUS reporter and transiently expressed in Nicotiana benthamiana leaf epidermal cells. The promoter activity of MdLBD3-pro-Del and MdG3OX3-pro-Del from ‘M9’ was significantly stronger than that of MdLBD3-pro-Ins and MdG3OX3-pro-Ins from ‘M9’ or ‘Malus baccata’, whilst the promoter activity of MdARF6-pro-Ins from ‘M9’ was lower than that of MdARF6-pro-Del from ‘Malus baccata’ or ‘M9’ (Fig. 7D).
To further examine the functional variations on the promoter sequences of these potential candidate genes, the promoter activity was re-examined by promoter truncation. Truncating off segments of promoter sequences, including the InDels of MdLBD3, MdARF6, and MdG3OX3 in ‘M9’, destroyed the promoter activity completely. The SNPs did not interfere with promoter activity, but the InDels on the each gene’s promoter altered the transcription activity significantly (Fig.7 A, B and D). Consequently, InDels on the promoter of MdLBD3, MdARF6, and MdG3OX3 were utilized as functional markers for dwarfing effects.
Motif analysis and transcription factor genes prediction
Full length of 1104 bp sequence of MdLBD3 promoter were analyzed through net software, revealing that the 11 bp deletion ‘TCAACAACATA’ located -339 bp from TSS weren’t broken TATA box or other motifs(Fig. 8A), but may have caused the generation of a new cis-acting element with the critical motif ‘TGGAATGGGTATAG’, which could be bound by some transcription factor of list with MPD numbers, but only MDP0000144203 was checked and validated confer to MD04G1244700 that was MdWRKY2 transcription factor (Fig. 8B).
Full length of 1289 bp sequence of MdARF6 promoter were analyzed through net software, revealing that the 14 bp insertion ‘GAGAGAGCGAGAGA’ located -954 bp from TSS was nearly to a GC box for dozens of bps(Fig. 8C), but the insertion had destroyed a motif ‘GGCGGTTGTG’ bound by the transcription factor of MDP0000147309 corresponding with MD07G1085000 that was MdGAMYB transcription factor (Fig. 8D).
Full length of 1176 bp sequence of MdG3OX3 promoter were analyzed through net software, revealing that the 10bp deletion ‘ATCATATTTT’ located -278 bp from TSS was in the middle of two TATA box (Fig. 8E), but may have caused the generation of a new cis-element with the critical motif ‘ACGTTTTTGT’, which could be bound by transcription factor of MDP0000144105 corresponding with MD12G1024200 that was MdABI5 transcription factor (Fig. 8F).
Functional analysis of allelic variation of the potential candidate genes
Prediction revealed that the 11 bp deletion of MdLBD3 may create a new cis-acting element could be bound by MdWRKY2 transcription factor, and Yeast one hybrid assay (Y1H) and co-expressing confirmed the binding of MdWRKY2 protein to the promoter of MdLBD3-Del in ‘M9’ but not to MdLBD3-Ins in ‘Malus baccata’ (Fig. 9B; Fig.7D). No differences in MdWRKY2 relative expression were identified between either ‘M9’ and ‘Malus baccata’ nor their F1 hybrids with dwarfing and non-dwarfing effects (Fig. 9A).
To the promoter of MdARF6 and it’s transcription factor MdGAMYB as predicted previous(Fig. 8C, D), by Y1H showed that the binding ability was completely absent between AD-MdGAMYB and BD-MdARF6-Ins (Fig. 9C), and the co-expressing revealed that the MdGAMYB protein could promote the activity of MdARF6-Del-GUS, but with no effect on MdARF6-Ins-GUS(Fig.7D), indicated that the 14 bp insertion specially on ‘M9’ broken the co-expressing between MdARF6-ins and MdGAMYB. Expression levels of MdGAMYB were quite analogous among ‘M9’, ‘Malus baccata’, and their F1 hybrids, irrespective of the dwarfing effects (Fig. 9A).
Y1H assay and co-expressing also confirmed that the 10 bp deletion on the MdG3OX3 promoter created a new cis-acting element on MdG3OX3 bind by MdABI5(Fig. 9D; Fig.7D), and the expression levels of MdABI5 were observed among ‘M9’, ‘Malus baccata’, and their F1 hybrids, irrespective of the dwarfing effects (Fig. 9A). These data suggested the use of the InDel variations MdLBD3, MdARF6, and MdG3OX3 as potential markers for dwarfing effects.
Prediction of dwarfing ability with marker genotype estimated genetic values (GEGV)
To estimate the genotype effects of the functional markers, MdLBD3(Del/Ins) /MdARF6(Del/Ins) /MdG3OX3 (Del/Ins) (abbreviated as Ld/Li, Ad/Ai, and Gd/Gi), 108 F1 hybrids of ‘Malus baccata’ × ‘M9’ were randomly chosen and genotyped. The marker genotype effects and effects of marker genotypic combinations on tree height, trunk diameter, and canopy width of the grafted trees were estimated (Table 3). AdAi had the largest marker genotype effect, and LdLi had the least (Table 3). The marker GEGV of the F1 hybrids were calculated by adding up the genotype effects of the three functional markers and the overall mean phenotype value. The Pearson’s correlation coefficients for tree height, trunk diameter, and canopy width between GEGV and observed phenotype value (OPV) were all significant (Fig. 10A, B and C). Five-fold cross validation produced an average correlation coefficient of r = 0.93 (P<0.01)(Fig. 10A, B and C), indicating a very high predictability of GEGV in the current hybrid population. To test the applicability of this GEBV in Malus germplasm, 64 Malus rootstock accessions were genotyped for Ld/Li, Ad/Ai, and Gd/Gi markers(Table 4). The GEGVs were consistent with the accession dwarfing ability reported by previous literature, implying that the GEGV model should be applicable for a broad spectrum of apple rootstocks (Table 4).
{kind=link}