Study sites
Entomological captures were carried out in 10 sites located in 8 municipalities in 5 departments of the country (Atlántida, Colón, Comayagua, El Paraíso, and Gracias a Dios) from February to October 2019 (Table 1). All the capture sites were located near small rural villages in which agricultural and fishing activities take place. Active foci of malaria were reported during 2018 at each site [8]. The departments of Atlántida, Colón and Gracias a Dios are classified as very humid tropical and coastal ecosystems with less than 550 m.a.l.s., while Comayagua and El Paraíso are considered as subtropical with heights above 550 m.a.l.s. and drier ecosystems. The average temperature varies between 25ºC and 33ºC, and the relative humidity ranges from 40% to 91% in all sites depending on the season of the year. The population’s livelihood in the selected areas is mainly based on agricultural and livestock activities. The study sites are those monitored by the Ministry of Health of Honduras to undertake routine entomological surveillance as they remain endemic to malaria by Plasmodium vivax. Malaria due to P. falciparum malaria is reported almost exclusively in Gracias a Dios. Geographical coordinates and altitude of the collection sites are shown in table 1.
Mosquito collection
Mosquito catches were performed for one night at each collection site. Atlántida and Colón were visited during the dry season of the year (February to April), and El Paraíso, Comayagua and Gracias a Dios were visited in the rainy season (August to October) (Table 1). Two collection methods were used at each site to capture the greatest amount and diversity of Anopheles species. The first method used outdoor CDC traps only fitted with light as an attractant, with 3 to 5 traps per site in a period from 18:00 pm to 6:00 am. Traps were separated a minimum of 50 meters from each other. Traps were placed in the outdoor structures of the houses where humans reside and also in structures where domestic animals rest. The second method was by aspiration of mosquitoes resting outdoors, during the period from 18:00 pm to 21:00 pm [17]. After collection, mosquitoes identified as anopheline were placed on a Petri dish with silica gel and transported at room temperature to the laboratory in Tegucigalpa where they were stored at -20°C until later morphological identification [18].
Morphological identification
After collection, all specimens were classified by sex and identified morphologically with a stereoscope using keys for anophelines of Central America and Mexico [19]. After morphological identification, wing and leg vouchers of each specimen were preserved as a reference in the Center for Genetic Research of the National Autonomous University of Honduras. Each mosquito was then stored individually at -20 °C for subsequent molecular tests.
COI gene
A subset of morphologically identified specimens were chosen randomly for molecular analysis. For the most common Anopheles species, between 8% and 17% of the specimens were selected to proportionally represent all the capture sites. For less common species, at least 50% of the specimens were selected for sequencing.
DNA was extracted from each specimen according to the protocol provided by the AxyPrep MAG Tissue-Blood gDNA Kit, Axygen® (Corning Incorporated, Life Sciences, Tewksbury, MA, USA). Preliminarily, the mosquitoes were macerated with a pestle in a 1.5 ml conical tube together with 50 µl of lysis solution provided by the kit. DNA was stored at −20°C until further use. Molecular analyses were performed on Anopheles mosquitoes to confirm species and calculate genetic variation within species. Two molecular markers were used: cytochrome c oxidase I gene (COI), and the internal transcribed spacer 2 (ITS2). The following primers were used to amplify a fragment of COI: LCO1490 GGTCAACAAATCATAAAGATATTGG and HCO2198 TAAACTTCAGGGTGACCAAAAATCA [20]. Reactions were carried out in a volume of 50 µl, with 25 µl of Taq Master Mix 2X (Promega, Madison, Wisconsin), 2.0 µl of each primer (10 µM), 2 µl of acetylated bovine albumin (BSA) (10 mg/ml), 4 µl of DNA, and nuclease-free water. The PCR program was as follows: 1 cycle at 95ºC for 10 minutes, 37 cycles at 94ºC for 1 minute, 48ºC for 1 minute, 72ºC for 1 minute, and 1 cycle at 72ºC for 7 minutes.
Some mosquito specimens that could not be amplified with the pair of primers described above were amplified using LCO1490 and a reverse primer described by Kumar et al [21]: AAAAATTTTAATTCCAGTTGGAACAGC (Fig. 1), with the following reagents and concentrations: 25 µl of Taq Master Mix 2X (Promega, Madison, Wisconsin), 1 µl of each primer (10 µM), 2 µl of DNA, and 21 µl of nuclease-free water. The cycling conditions were: 1 cycle at 95ºC for 5 minutes, 5 cycles at 94ºC for 40 s, 45ºC for 1 minute, 72ºC for 1 minute, 37 cycles at 94ºC for 1 minute, 54ºC for 1 minute, 72ºC for 90 s, and a final extension at 72ºC for 10 minutes. The PCR products were separated by electrophoresis in 1% agarose gels with ethidium bromide.
ITS2 ribosomal region
For ITS2 amplification, PCR reactions were performed using the universal primers [22]: 5.8S ATCACTCGGCTCGTGGATCG and 28S ATGCTTAAATTTAGGGGGTAGTC. Reagent concentrations were as follows: 25 µl of Taq Master Mix 2X (Promega, Madison, Wisconsin), 2 µl of each primer 10 µM, 2 µl of DNA, and water for a total reaction volume of 50 µl. PCR amplifications were performed with the following conditions: 94ºC for 2 min, 34 cycles of 94ºC at 30 s, 57ºC at 30 s, 72ºC at 30 s, and final extension of 72ºC at 10 min.
Sequence analysis
The amplification products of both COI and ITS2 markers were sequenced on both strands using the same primers of the PCR. A representative subset of mosquitoes of all species and all collection sites was selected for sequencing. Purification and sequencing services were provided by Psomagen (https://www.macrogenusa.com). The sequences were edited with the Geneious® 9.1.7 software (Biomatters Ltd. Auckland, New Zealand) and were deposited in two databases: Barcode of Life Data System (BOLDSYSTEMS, http://www.boldsystems.org), and in the National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov). Barcode Index Numbers (BINs) and accession numbers were obtained for each sequence. All sequences were submitted as queries to NCBI through the BLAST tool [23] under default parameters to identify the most similar sequences in the GenBank nucleotide collection.
Nucleotide diversity (π) and number of haplotypes
In order to calculate the nucleotide diversity (π), the sequences of both molecular markers were analysed separately and by species. The sequences were aligned using the MUSCLE algorithm. MEGA v10.0 software with 1000 Bootstrap replicates was used to calculate the pairwise distance using the Maximum Composite Likelihood substitution method, and 95% as the site coverage cut-off. The percentage of identical bases for each species and between species was calculated in order to demonstrate the reported “barcoding gap”, which is the difference between inter- and intraspecific genetic distances within a group of organisms.
The haplotype diversity was calculated with R through the function hap.div of pegas (v0.12 package) and using the Nei and Tajima´s method [24]. Haplotype frequencies were calculated using the Haplotype function with default parameters, and the haplotype network was computed with the haploNet function using an infinite site model, pairwise deletion missing data, and probability of parsimonious link [25].
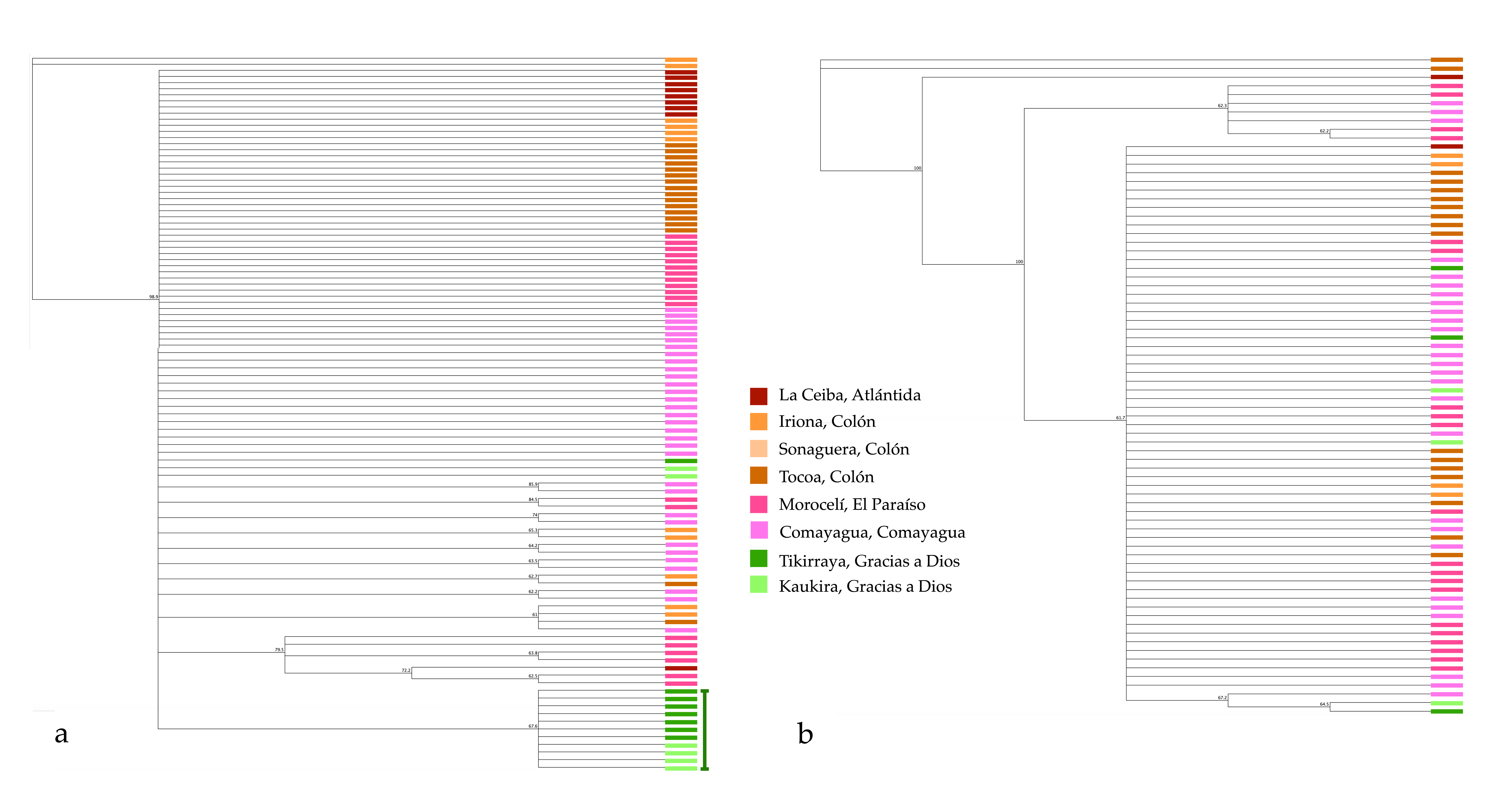
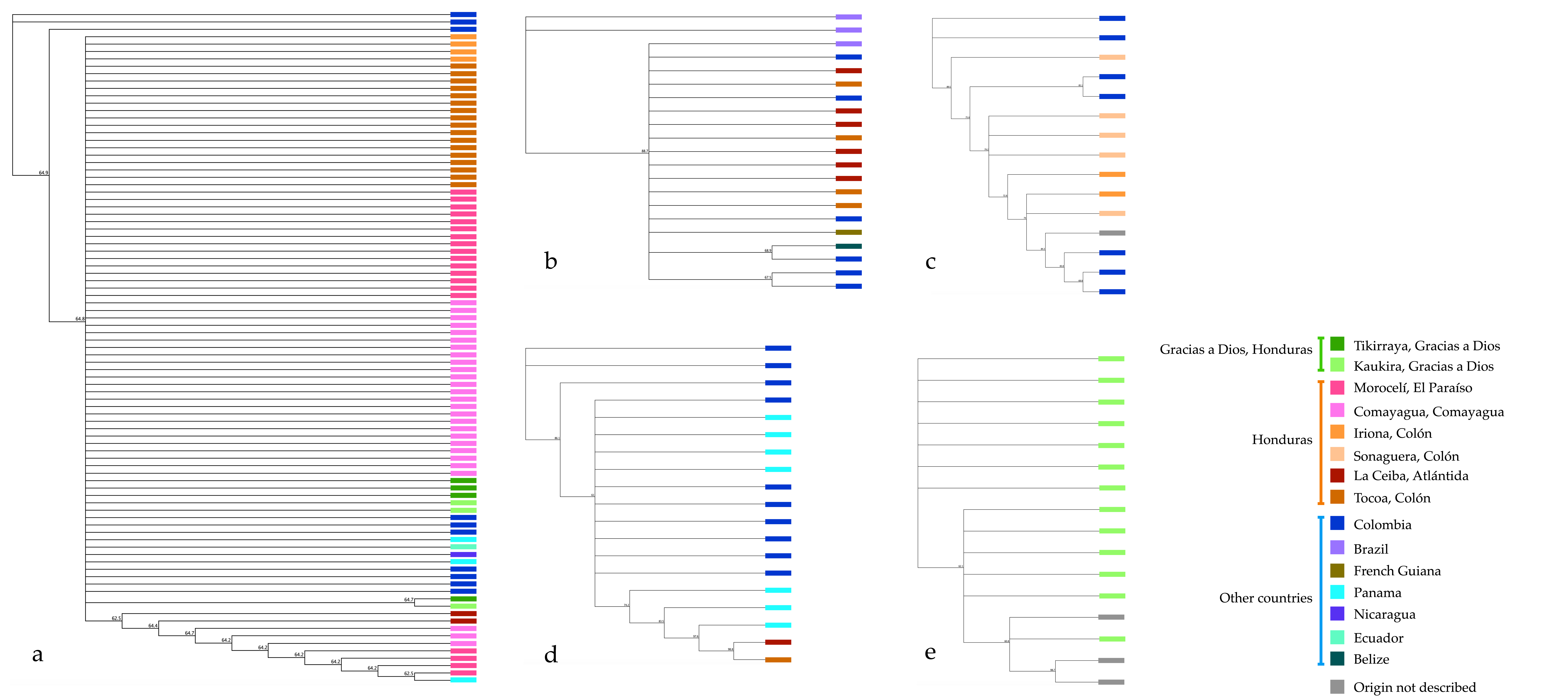
Phylogenetic analysis
Nucleotide sequences were trimmed and manually corrected using the Geneious® 9.1.2 software (https://www.geneious.com). The ClustalW tool was used to align sequences. Phylogenetic trees were constructed using the Tamura-Nei distance model, the Neighbor-Joining method and a bootstrap of 1,000 replicates with no outgroup. Length, identical sites and pairwise % identity were calculated for each molecular marker and each species.
To calculate the phylogenetic relationships between specimens collected in Honduras with those collected in other countries of the Americas, analogous COI and ITS2 sequences for all available Anopheles species were downloaded from the GenBank database. Sequences were aligned and phylogenetic trees constructed under the same parameters described above.
{kind=link}
{kind=link}