The S protein of coronavirus directly interacts with cellular receptors, the diversity of the S gene is often used as the phylogenetic marker in evolutionary analysis. The complete genomes of diverse strains to the global database promotes better understanding of evolutionary and phylogenetic relationships [14]. In the study of virus evolution, one method of testing for selection is to compute the ratio of nonsynonymous to synonymous substitution rates (ω), ω is expected to have a value of 1 under the assumption of neutral evolution. Positive and negative selection are indicated when ω > 1 and ω < 1, respectively [23]. The M8-M7 comparison model offers a very stringent test of positive selection [1]. In terms of S gene of different coronavirus, M8 model was a better fit than the M7 model, the favor of positive selection here and the high mutation rate of the S gene [13] make it the best target for evolutionary assays, which also was confirmed in the evolutionary comparison of the whole genome sequence with the S gene and other structural protein genes.
Previous study showed that the natural selection was the main force influencing the codon usage pattern of PEDV, while mutation pressure played a minor role [6, 41, 44]. Here, if positive selection is the driving force for the higher synonymous substation rate seen in spike, we expect the FOP of spike to be different from that of genome, the elevated synonymous substitution rate measured in S gene might be more likely caused by higher mutational rates, but the FOP of S gene showed no obvious difference with that of the genomic average, the underlying molecular mechanism remains unclear, deserving further studies. Synonymous substitutions may serve as another layer of genetic regulation, guiding the efficiency of mRNA translation by changing codon usage.
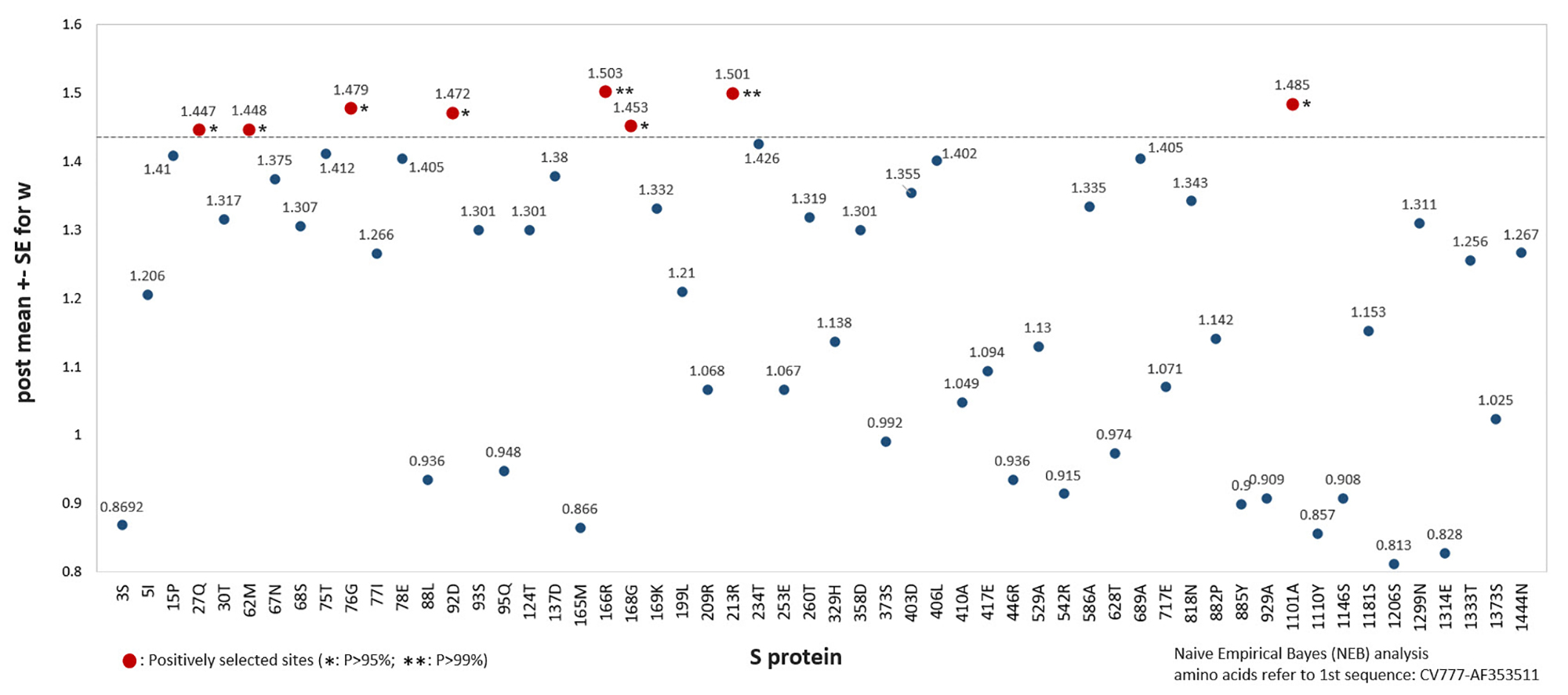
By the application of NEB analysis, we found that S and N protein were favor of mutation than other proteins in this study, which was consistent with antigenic study of possible antigenic differences emerged in both the spike and nucleocapsid proteins between different genogroups [16]. As we showed here, the S gene was considered to provide the maximal interpretative power in PEDV evolution for its highest phylogenetic signal with substitution rate and phylogenetic topology similar to those obtained from the complete genome [34, 35], which would facilitate the data analysis more lightly.
Variations in the S protein are important for revealing the genetic evolution and the pathogenicity of PEDV strains [11, 33, 39]. Structurally, we found that the highly mutated residues of S protein were S1 domain dominated, indicating a strong S1-related evolutionary pattern of PEDV, which might be induced by the antigenic drift as amino acid positions with significant variation among isolates from different regions and subgroups were found [13]. PEDV S protein may undergo a conformational change after receptor binding and cleavage by exogenous trypsin, which induces membrane fusion [24].
Selective pressures drive adaptive changes in the coronavirus S proteins directing virus-cell entry. The high hypervariability in the SARS-CoV-2 S protein appears to be driven by counterbalancing pressures for effective virus-cell entry and durable extracellular virus infectivity [27], which could be caused by the variation of amino acid positions. The binding domain of the PEDV cellular receptor APN was shown to reside within a domain in the C-terminal of S1 domain (residues 477–629), which is closed to one of the sites we found in haplotype networks for the potential differentiation of PEDV genotype. The strain of TW/Yunlin550/2018 (MK673545.1) that located on the edge of the genotypes of GI and GII showed a completely different pattern in the 6 nt site (AATAGA, aa of Asn-Arg, NR) compared with the GI strains (GGAAAA, aa of Gly-Lys, GK), which might represent a consequence for a better host adaption during virus evolution when facing the counterbalancing pressure, a further study for the evolution pattern is needed.
Geographically, the GI strains of Europe were evolutionarily separated from the GII strains in other global regions. In the genotype GII, the America strains were clustered with most of the strains in Japan and South Korea, and then strains in China are shown in a more compact branch. The increasingly international pig industry involves the trade of various breeding materials and animals, which may bring the risk of disease transmission. The global exchange of ingredients has created demand for products that prevent disease transmission from the feed, such as the use of the monoglyceride blend could mitigate and prevent PED transmission in piglets from contaminated feed [26].
Overall, we found that S protein showed strong signals of positive selection, and it is more representative of the evolutionary relationship at the genome-wide level than other genes. Structurally, the evolutionary pattern of S protein is highly S1 domain related, which also represents the marker for clustering lineages corresponding to genotype GI and GII geographically. These findings provide several fundamental insights into the evolution of PEDV and the guidance for developing effective prevention countermeasures against PEDV.
{kind=link}