Cell culture
HepG2 (ATCC® HB-8065™) and Huh-7 cells were routinely cultured at 37˚C with 5% CO2 in GlutaMAX™ supplemented, high glucose Dulbecco modified eagle medium (DMEM) (31966047, ThermoFisher Scientific, Stockholm, Sweden) with 1% antibiotic antimycotic solution (A5955-100ML, Sigma-Aldrich, Darmstadt, Germany) and 10% fetal bovine serum (FBS) (10270106, ThermoFisher Scientific, Stockholm, Sweden). No FBS was used during starvation. Misidentification of cell lines were checked at the Register of Misidentified Cell Lines and chosen cell lines were not on the list. Extracted DNA from all cell lines is sent yearly to Eurofins Genomics (Ebersberg, Germany) for cell line authentication using DNA/STR-profiles. Authentication confirmed the correct identity of our cell lines and tested negative for mycoplasma contamination.
For 2D cell culture, a HepG2 and Huh7 single cell suspension was prepared by trypsinization and seeded into 96-well plates at a seeding density of 1.5 x 104 cells/ml, with each well containing 3000 cells in 200µl culture medium. For 3D cell culture a modified setup of cells embedded into a hydrogel consisting of fibrinogen (From bovine plasma, F8630-5G, Sigma-Aldrich, Darmstadt, Germany) and collagen (Rat-tail type I, 50201, Ibidi, Lund, Sweden) were prepared as previously described8 (table 2).
Briefly, a stock solution of fibrinogen (60 mg/ml) was prepared. The HepG2 or Huh7 cells were prepared as single cell suspensions, counted, and diluted to 2 x106 cells/ml. The diluted cell suspension was centrifuged at 300 xg for 3 min, and the supernatant removed. Hydrogels were added to the cells and 200 µl of the gel was seeded onto 12-well plates. Crosslinking of the gels was performed in the culture hood for 15 min at room temperature, followed by 45 min at 37°C within a CO2 incubator. Growth medium was subsequently added to each well and replenished every second day. Gels were maintained for 21 days prior to experimentation, unless stated otherwise.
Animal study
Five-week-old male sv129-mice were injected every other week with 35 mg/kg diethyl nitrosamine (DEN) or equal volumes of saline. In this model, tumours occur after 25 weeks, and mice livers were sampled post-mortem after 28 weeks 18. All methods were approved by the Uppsala Ethical Committee for Animal Experimentation (DNR 5.8.18-0089/2020).
Rheology
All measurements were done in triplicate. Hydrogels were measured using a Discovery Hybrid Rheometer 2 (TA instruments, Sollentuna, Sweden). All measurements were performed at a frequency of 1.1 Hz and Frequency sweeps from 0.02-2 Hz 0.267% shear strain to study the differences in storage moduli an indication of stiffness. The hydrogels were measured on day 1 of preparation, 11 and 21, both with and without cells to determine any potential cell mediated remodelling. For mouse liver samples, livers were collected from the left liver lobe using an 8mm and biopsy punch. Samples were submerged in PBS and kept on ice, until rheology measurements. All measurements were performed at 37°C, with a constant axial force of 0.1N using an 8 mm diameter parallel plate stainless steel geometry.
Resazurin reduction assay
Viability and proliferation were measured using a Resazurin reduction assay, following manufacturer´s guidelines. For 2D cultures culture medium was removed and cells were washed twice with 200µl PBS. Starvation medium was added, and cells were incubated for 2-hours. Following incubation, starvation medium was removed and 200µl of the Resazurin solution was added to the cells and incubated overnight. For 3D cultures, culture medium from the gels were removed, and the cells washed twice with PBS and 1ml starvation medium was added. After a 2-hour incubation at 37°C, starvation medium was removed and 1 ml Resazurin solution was added to each well, followed by overnight incubation at 37°C. Following incubation 200 µL was transferred from each well into a Corning black clear bottom 96-well plate (Sigma-Aldrich, Darmstadt, Germany). Fluorescence was read using a FLUOstar Omega microplate reader (BMG Labtech) at excitation and emission wavelengths of 485 and 550 nm, respectively.
Collagenase-dispase cell isolation
To isolate cells from hydrogel formulations, cell culture medium was removed, and the gels washed twice with 1 ml PBS. A volume of 700µl collagenase-dispase (1mg/ml) in PBS, was added to each gel, and the gels were incubated at 37°C for 90 min. To assist with the dissociation step, gels were manually disrupted with a pipette at 20 min intervals. Once the gel was completely digested, the reaction was inactivated by adding 30µl EDTA (10mM). The resulting cell suspension was collected and centrifuged for 15 min at 1000 xg. Following centrifugation, supernatant was removed, and cell pellet was resuspended in lysis buffer or culture medium depending on the downstream application.
Protein content
Protein content was determined using the Pierce™ BCA Protein Assay kit (23225, ThermoFisher, Stockholm, Sweden), according to manufacturer's recommendations. Cells from hydrogels were isolated using collagenase-dispase and transferred to 2ml collection tubes. Cell suspensions were subsequently centrifuged for 5 min at 500 xg. Culture medium was aspirated, and cells were washed twice in PBS followed by lysis in 300 µl RIPA buffer supplemented with protease inhibitors for 20 min on ice. Samples were then centrifugated at maximum speed for 10 min and the supernatant was collected and transferred to a clear flat bottom 96-well plate.
Albumin Enzyme-Linked immune Sorbent Assay (ELISA) to determine functionality
Cell culture medium from 3D hydrogel cultures were collected on day 1, 11 and 21. Samples were centrifuged for 2 min at 5000 xg and the supernatant was transferred to a new collection tube and frozen. Albumin levels were measured by means of the Human Albumin (ALB) ELISA kit (EHALB, ThermoFisher, Stockholm, Sweden), following manufacturer´s guidelines with a 1:500 dilution of sample. Urea levels were measured by means of the Urea Nitrogen (BUN) colorimetric detection kit (EIABUN, ThermoFisher, Stockholm, Sweden), following manufacturer´s guidelines but without sample dilution. The averages from three biological replicates were used for calculations. All data was normalised to total protein content.
Drug response and experimental group setup
The response to DOX (1µM), Sorafenib (10µM) and NITA (10µM) was determined in the 2D-setup and hydrogel formulations. HepG2 and Huh7 cells were seeded onto black flat clear bottom plates and left overnight to attach. Three-dimensional hydrogel formulations were prepared and maintained for 21-days. Prior to drug treatment, culture medium was removed and both 2D and 3D cultures were washed twice with PBS. Starvation medium was added for 2 hours, and cultures were subsequently incubated at 37°C for 2 hours. Following incubation, starvation medium was removed, and cultures were treated with DOX, sorafenib and NITA in low glucose medium (3mmol/L) and pH 6 for 96hrs. All measurements were done in triplicate.
Drug distribution and permeation study
The permeability of DOX through the 3D cell model was assessed using an experiment inspired by an established protocol 51. Cell culture media buffered with 25 mM HEPES (pH=7.4) was used to maintain both donor and acceptor side pH. DOX (1 mM) was added to the donor side (insert) of the model. Samples (100 µL) were taken every 30 min from the receiver side (well) and an equivalent volume of fresh cell media was added (i.e., sample & replace method). The 12-well plates were gently agitated (100 rpm) and kept at 37°C throughout the experiment, except for when sampling at room temperature. After 2hr the permeability experiment was ended, the donor side sampled (50 µL), the hydrogel washed with PBS and collected using a spatula and the receiver media pH measured. DOX and its main metabolite doxorubicinol (DOXol) were quantified using a previously published UPLC-MS method 52. Briefly, the analytes of interest were extracted overnight from receiver side samples, as well as from the collected hydrogels, using protein precipitation with ice-cold acetonitrile containing set levels of isotopically labelled internal standards (1 µM). Donor side samples were diluted by a ten-fold in cell culture media before being treated similarly to receiver side samples. Data were processed in TargetLynx (MassLynx V4.1, Waters Corporation, Milford, MA, USA) using linear curve fitting (weighting factor of 1/x) of the peak area ratio (analyte:internal standard) as a function of the analyte concentration. Linear calibration curves (R2>0.99) in cell culture media for both DOX and DOXol were constructed between 0.125 and 25 µM and the lowest point in the calibration curve (125 nM) corresponded to the lowest limit of quantification (LLOQ). Apparent permeability coefficients (Papp, cm/s) were calculated according to previously described equations 53. Mass balance (%) was calculated based on donor and receiver amounts. DOX and DOXol hydrogel concentrations (nmol/g) were calculated by dividing the quantified extracted amount (nmol) from the gel by the weight of the collected gel (g).
Plannimetry
Photomicrographs were taken of spheroid formation in hydrogels containing HepG2 cultures on day 14 and 21 using an Olympus IX81 motorized microscope and an Olympus DP71 camera. A total of 150 spheroids were measured in each condition at both time points using Fiji Image J software.
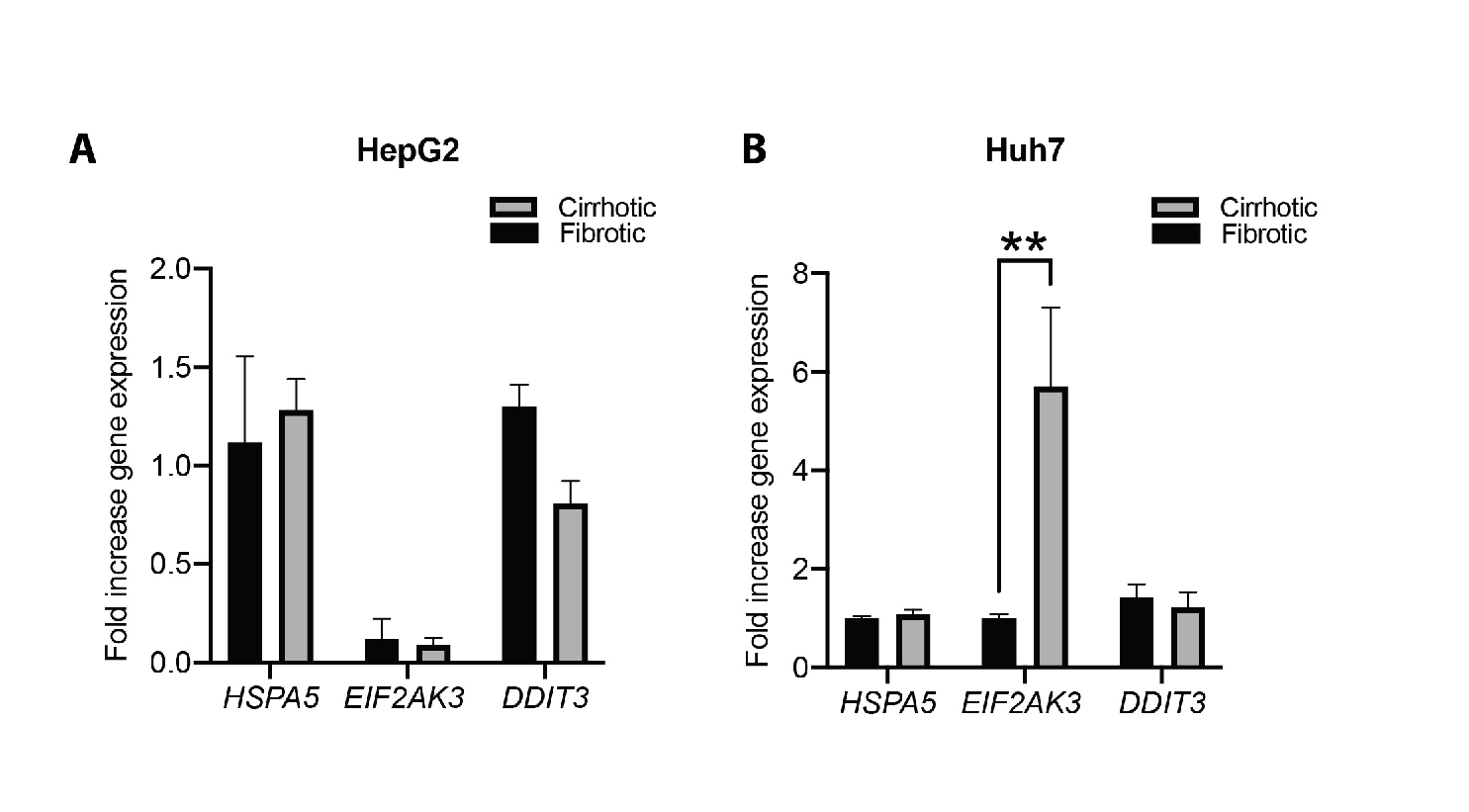
Quantitative RT-PCR
RNA was isolated using the EZNA® RNA isolation Kit II (R6934-02, VWR, Spånga, Sweden). RNA-concentration and purity were evaluated using Nanodrop, followed by reverse transcription of 300ng mRNA using the iScript select cDNA synthesis kit (1708897, Bio-rad, Solna, Sweden). Amplifications were done using primers summarized in table 3. In all instances, mRNA-expression was normalized to housekeeping gene GAPDH, using the delta-delta-CT method to calculate fold change of three biological replicates.
Statistics
Unpaired, two-tailed Student’s T-test or one-way analysis of variance (ANOVA) followed by Tukey´s multiple comparison test was performed using GraphPad Prism version 8.0 to determine statistical significance. P-values <0.05 were considered statistically significant. Experiments were done in at least three biological replicates, which we define as parallel measurements of biologically distinct samples taken from independent experiments. Technical replicates we define as loading the same sample multiple times on the final assay. Outliers were kept in the analyses, unless they were suspected to occur due to technical errors, in which case the experiment was repeated.
{kind=link}