Urine collection and processing
Urine samples from forty healthy individuals (aged 20–72 years) were collected from Apollo Hospital, Hyderabad. Demographic characteristics are represented in Table 1.
Table 1
– Healthy control demographics
|
Parameter
|
value
|
|
Sample Size
|
40
|
|
Age
|
47.2 ± 13.4
|
|
Gender
|
|
|
Male
|
35
|
|
Female
|
5
|
Table 1: Urine samples were collected from 40 healthy individuals whose age is represented as Mean ± SD.
Urine was collected in collection containers, and 4.2ml protease inhibitor (1mg/ml Leupeptin (G Biosciences, MO, USA), 10mM Sodium Azide, 50mM PMSF ( both from Sigma, MO, USA) per 50ml of urine was added. Before proceeding with UEV isolation, all urine samples were subjected to urine tests using the urinalysis strips (Siemens Multistix 10 SG, Siemens, Munich, Germany). The urinalysis strips were dipped into the urine containing tube, and excess urine was drained. The colour changes on the strips were observed and quantified using Siemens instrument CLINITEK Advantus for the parameters shown in Table 2.
Table 2
– Summary of urine analysis of healthy individuals
|
Samples (40)
|
Leukocyte
|
Nitrite
|
Urobilinogen (mg/dl)
|
Protein (mg/dl)
|
pH
|
Blood
|
Specific Gravity
|
Ketone
(mg/dl)
|
Bilirubin
|
Glucose
(mg/dl)
|
Colour
|
Clarity
|
|
Observed
|
Negative
|
Negative
|
0.2
|
Negative
|
5.5–7.5
|
Negative
|
1-1.03
|
Negative
|
Negative
|
Negative
|
Yellow
|
Clear
|
|
Reference range
|
Negative
|
Negative
|
0.2–3.2
|
Negative
|
5-8.5
|
Negative
|
1-1.03
|
Negative
|
Negative
|
Negative
|
|
|
The collected urine samples were immediately processed for UEV isolation using the modified salt precipitation method described previously (5). Briefly, 30–50 ml of first void urine samples were centrifuged at 1800*g for 10 min at 4°C to clarify the cell and cellular debris. The cell pellet was washed with 1x PBS (Hyclone GE Healthcare, IL, USA) and resuspended in RNA later solution (Sigma) for RNA isolation. The supernatant was carefully transferred to a fresh tube containing 20mMTris-EDTA (Sigma) pH 6.8, and the pH was adjusted to 4. The supernatant mixture was vortexed for 90sec and further centrifuged at 8000*g for 15min at 4°C. After the centrifugation step, the supernatant was collected in a fresh tube, an equal volume of 2x PEG solution was added (final 12% PEG in 0.5M NaCl (Sisco Research Laboratories (SRL), Mumbai, India) and mixed thoroughly. The UEV pellet obtained was then treated with 500µl of 100mM DTT (Sigma) and incubated for 10 min at 37°C for Tamm-Horsfall Glycoprotein (THP) removal. Subsequently, the DTT treated pellet was centrifuged at 17000*g for 15 min at 4°C with zero brakes. The supernatant obtained here was added to the previously derived PEG- supernatant mixture and incubated overnight at 4°C. The next day, the PEG-supernatant mixture was centrifuged at 10,000*g for 60 min at 4°C. Finally, the pellet containing UEVs was resuspended in 1x PBS and stored at -80°C for further use.
Urinary Extracellular Vesicle Characterisation
The isolated UEVs were subjected to biophysical and biochemical characterization to ensure the quality of isolated vesicles.
Morphological characterization
TEM Analysis
Transmission electron microscopic analysis was performed to examine the size and confirm the presence of intact exosomes and size determination. Briefly, UEV samples were fixed with 1% glutaraldehyde for 5 min on the 400 mesh copper grids (FCF400-Cu, Electron Microscopy Sciences, Hatfield, PA). The grid was washed twice with water, and further stained with 2% uranyl acetate, and then air-dried at room temperature. Images of the isolated UEVs were captured using a transmission electron microscope (JEM-2100, JEOL Ltd. Tokyo, Japan).
Nanoparticle Tracking Analysis
Nanoparticle tracking analysis is a powerful technique that utilizes laser light scattering microscopy combined with Brownian motion to determine the size, concentration, and morphology of extracellular vesicles [45]. We performed NTA on the isolated UEVs using NanoSight LM10 instrument (NanoSight, Amesbury, UK), where the laser light-scattering was measured at 488 nm, and the relative concentration and size was determined.
Dynamic Light Scattering
The size of UEVs was further determined using the dynamic light scattering (DLS) technique that measures the size based on the Brownian motion of the dispersed particle as described previously[46]. 3ml homogenous suspension of UEVs (diluted 1:100 in 1X PBS) was transferred to a cuvette, and the hydrodynamic diameter of exosomes was measured in a DLS instrument (Nicomp Z3000, Entegris, MA, USA).
Biochemical Characterisation
Protein estimation and SDS-PAGE analysis
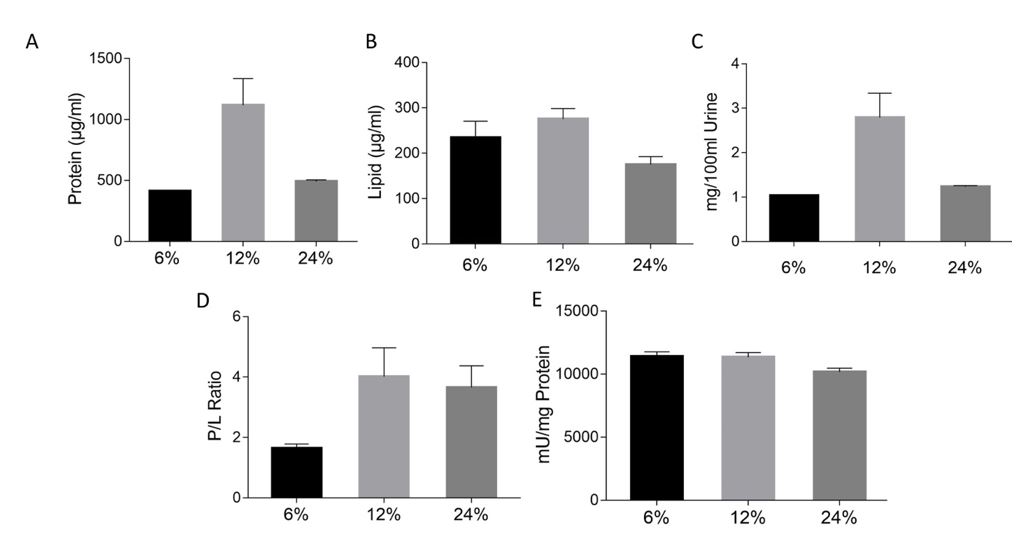
The total protein content in isolated UEVs was quantified using a bicinchoninic (BCA) protein assay kit (G-Biosciences) following the manufacturer's instructions. Samples were stored at -80°C until further analysis. For SDS-PAGE, 10µg of UEV protein was lysed using RIPA lysis and extraction buffer (G-Biosciences) including protease inhibitor cocktail (Roche, Basel, Switzerland) followed by incubation at 4°C for 15min. The lysed protein samples were then mixed with reducing sample buffer and denatured for 10 mins at 70°C. The exosomal proteins were resolved on 10% SDS PAGE for 1.5 h at 120 V, and the gels were stained using 2% silver nitrate.
Lipid Estimation
Total lipid content of the UEVs was quantified using Phospho Vanillin Assay, as reported previously (35). Briefly, 200µl of 96% H2SO4 was added to 40µl of the lipid standard, DOPC (1,2-Dioleoyl-sn-glycero-3-phosphocholine), or the UEV samples and evaporated at 90°C on a dry bath for 20 min. After the tubes were cooled to RT, 120µl of Phosphovanillin reagent (200µg vanillin in 17% H3PO4) was added. The reaction is allowed to occur by incubating for 1 h at 37°C, and then absorbance was recorded at 540nm.
AChE activity
Acetylcholinesterase (AChE) activity assay was carried out to confirm the presence of acetylcholinesterase (Sigma), which is considered as a marker enzyme for exosomes[21].20ul UEV fraction was added to 96-well flat-bottomed microplate. Acetylthiocholine iodide (final 1.25mM) and 5’, 5’-dithio-bis (2-nitrobezoic acid) (final 0.1mM) (both from Sigma) was then added to each well at a final volume of 300ul. The absorbance was recorded at 412nm every 5 min for 30 min. The amount of AChE activity in the exosome fraction was determined from the AChE enzyme's standard curve.
Dipeptidyl peptidase-IV activity
Dipeptidyl peptidase-IV (DPPIV), a membrane-associated peptidase, is associated with kidney diseases and is secreted by tubular epithelial cells in the kidney. Hence, it is considered to be a component of urinary microvesicles in the urine. We used a colorimetric assay previously used to determine serum DPPIV activity by quantifying DPPIV in UEVs as additional criteria to assess exosome quality[31]. Briefly, 10µl of UEV sample was mixed with 50µl of 71mmol/l glycine/NaOH (pH 8.3) buffer in 96 well plate. 50µl of 0.5mg/ml substrate Gly-Pro-p-nitroanilide (Sigma) was added to all wells, including blank wells, and incubated for 60 min at 37°C. The substrate is cleaved by DPPIV in the sample which releases free 4-nitroaniline, a chromogenic, whose absorbance is measured at 405nm in a plate reader. DPPIV activity in the sample was calculated against the standard plot of p-nitroaniline standard (Sigma).
Antibody Array
Exo-Check exosome antibody array (SBI, Systems Biosciences, USA ) was performed according to the manufacturer's instructions. The array contains 8 antibodies for known exosome markers including (CD63, CD81, ALIX, FLOT1, ICAM1, EpCam, AnXA5, and TSG101) and 4 controls, including two positive controls, blank and gm130 cis-Golgi marker which monitors for any cellular contamination. Briefly, 100µg of UEV protein was treated with lysis buffer; 1µl of Labelling reagent was then added and incubated at RT for 30mins with constant mixing. The column provided in the kit was equilibrated with column buffer before the sample was eluted. The eluted lysate was mixed with blocking buffer, transferred on to membrane, and incubated at 4°C overnight on a rocker. The next day, the membrane was washed thrice (5 mins each) with washing buffer and incubated with detection buffer for 30mins at RT on a shaker. The membrane was washed thrice again with washing buffer before development. Super Signal West Femto Maximum sensitivity substrate (Thermo Fisher Scientific, US) was used for chemiluminescence detection of the membrane in the imaging system (ChemiDoc, BioRad, USA).
RNA isolation and quantification
Total RNA from UEV was isolated using TRIzol™ LS Reagent (Invitrogen, California, USA) following the manufacturer's instructions. Briefly, the exosome sample was mixed with Trizol Reagent (1:3) and incubated for 5 min at RT. Chloroform was added (1:5) to the exosomal mixture and incubated for 15 min at RT, followed by centrifugation at 12000g for 15 min at 4˚C. The aqueous phase was carefully removed and transferred to a fresh tube. To all the tubes equal volume of isopropanol, 500µg/ml Glycogen (GlycoBlue™ co-precipitant Invitrogen), and 0.5µg yeast tRNA (Invitrogen) was added and incubated overnight at -20˚C. The samples were then centrifuged at 12000g for 15 min at 4˚C, followed by an ethanol wash. Finally, the air-dried RNA pellet was resolved in nuclease-free water and stored at -80°C for further analysis. The RNA content was determined using Qubit™ RNA HS Assay Kit (Invitrogen). Additionally, the UEV RNA quality and quantity were analysed in Agilent Bioanalyzer 2.1 instrument using RNA Pico kit for high sensitivity analysis of total RNA and mRNA samples (Agilent Technologies, California, USA).
Reverse Transcription
Exosomal RNA was reverse transcribed with High capacity cDNA Reverse Transcription kit (Invitrogen) using 10µL exosomal RNA and 10µl cDNA master mix prepared according to the manufacturer's instruction. cDNA was pre-amplified using Sapphire Amp fast PCR master mix (Takara Bio Inc. Shiga Prefecture, Japan) Pre-amplification with pooled primers was carried out for 20 cycles at 95˚C for 5 min, 95˚C for 1 min, 60˚C for 30 sec, 72˚C for 1 min, and 72˚C for 5 min and hold 4˚C. The pre-amplified transcript was then used as a template for PCR and RT PCR analysis.
Endogenous gene expression in Urinary EV
In the present study, we selected 5 reference genes, namely GAPDH, B2M, RPL13A, PPIA, and HMBS, for normalization of quantitative real time PCR in UEV samples from healthy individuals. All primer sequences were custom synthesized by the supplier (Bio serve, India), a detail of which is listed in Table 3. The pre-amplified samples mentioned in the above section were diluted 1:5 with nuclease-free water, and 1µl of the diluted product was used as a template for semi-quantitative as well as quantitative PCR. Briefly, a 10µl reaction was performed in Applied Biosystem 7500 Real-Time System using 5µl TB Green Premix Ex Taq (Takara Bio Inc.), 0.1µl 10µM primer pair, 1µl template cDNA, and 3.8µl nuclease-free water. Real-time PCR was conducted for 30s at 95˚C, followed by 35 cycles of 5s at 95˚C and 30s at 60˚C. All samples were evaluated in duplicates, and for every gene analyzed, a non-template control (NTC) was also included. PCR product purity was monitored from melting curve analysis and 2.0% agarose gel electrophoresis.
Table 3 - Primer pair sequences of the candidate reference genes used in this study.

Normalization of housekeeping genes
To analyze the candidate gene expression stability, normalization analysis was performed using online available software RefFinder. RefFinder determines the best stably expressed candidate reference gene by generating a comprehensive ranking for each gene based on the combined expression stability data from four statistical algorithms- Genorm, Normfinder, Bestkeeper, and Delta Ct. These mathematical algorithms use threshold cycle (Ct) values to calculate expression stability of each candidate reference gene. Precisely, Normfinder implements an ANOVA based algorithm to determine inter and intragroup variations in the reference gene expression, which is represented in terms of stability value. The gene expression stability score in Genorm is determined by pairwise comparison, while Bestkeeper applies pairwise correlation analysis. Delta Ct method calculates variations in delta Ct by comparing relative transcription of pairs of genes. Ranking of genes was done based on the stability score, where genes with the lowest stability score were considered more stable.
Statistics
For the comparative analysis of exosomal characteristics using the PEG vs. Kit method, a paired student's t-test was done to test the statistical significance using GraphPad software version 8.3 was used. Only P < 0.05 was considered statistically significant. All experimental data are shown as mean ± SEM unless otherwise mentioned. Gene stability analysis of housekeeping genes was performed in RefFinder. The stability value or SD data obtained using different algorithms were used for the ranking of genes.
{kind=link}