Primary bronchial epithelial cells and BEAS-2B cells: Primary bronchial epithelial were isolated and characterised as described earlier [20]. Endo-bronchial tissue biopsies were obtained from six patients by endo-bronchoscopy for diagnostic reasons by the lung clinic (University Hospital Basel, Switzerland). All patients provided written informed consent for the use of one additional anonymised biopsy for scientific investigations. The study was approved by the local Institutional Ethical Committee (EKBB 05/06).
Epithelial cells were isolated by cell-type selective medium CnT-PR-A (CellnTec, Bern, Switzerland). BEAS-2B cells were grown in the same cell culture medium. The cell phenotype was monitored by phase contrast microscopy and by staining for: positive E-cadherin and cytokeratin-13, as well as for negative fibronectin staining (Fig. 1a).
Drugs: EPs® 7630, a herbal drug preparation from the roots of Pelagonium sidoides (1:8–10), extraction solvent: ethanol 11% (w/w), supplied by Schwabe Pharma AG (6403 Küssnacht am Rigi, Switzerland) was dissolved in cell culture medium.
Vitamin D, calcitriol (Sigma-Aldrich Merck, Buchs, Switzerland), was dissolved in ethanol (100 µg/ml) before being added to the cell cultures at final concentrations ranging from concentrations of 0.1–10 µM, together with EPs® 7630.
High Pressure Liquid Chromatography: EPs® 7630 high pressure liquid chromatography -UV High Resolution Mass Spectrometry (HRMS): The HPLC-UV-HRMS chromatograms were recorded on an Thermo® Vanquish UHPLC coupled to an DAD and Thermo Orbitrap® Fusion mass detector using a Waters® Atlantis T3 (3 µM, 2 x 150 mm) column without pre-column. Eluent A consisted of 2.5% (v/v) acetonitrile and 0.5% (v/v) formic acid in water. Eluent B consisted of 5% (v/v) water and 0.5% (v/v) formic acid in acetonitrile.
At a flow rate of 0.2 mL/min, the gradient was as follows: from 0.0–10.0 min. linear for 0–5% eluent B, from 10.0–65.0 min. linear for 5–50% eluent B, from 65.0 to 66.0 min. linear for 50–100% eluent B, from 66.0 to 71.0 min. isocratic 100% eluent B column wash, from 71.0–72.0 min. linear from 100–0% eluent B followed by 8 min. equilibration period with 0% eluent B, resulting in a total run time of 80.00 min. UV detection wavelength of 280 nm and a column temperature of 40 ºC were applied. The injection volume was 4 µL of a 5 mg/mL Pelargonium sidoides extract EPs® 7630 dissolved in eluent A. HRMS based peak assignment was performed using ACDLabs Spectrus Processor Software v2017.2.1.
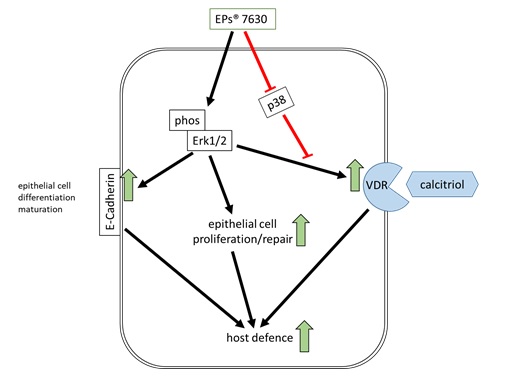
Cell treatment: Confluent epithelial cells were stimulated with EPs® 7630 (0.1–10 µg/ml), or calcitriol, or the combination of both, for up to 48 hours before being infected with 1 MOI of RV-16 as depicted in Fig. 1b.
For EPs® 7630-induced VDR expression, cells were pre-treated for 24 or 48 hours with EPs® 7630 (10 µg/ml). To determine the effect of EPs® 7630 and calcitriol on VDR translocation pre-treated cells were exposed to increasing concentration of vitamin D (calcitriol 0.1–10 µM in DMSO). The expression of the VDR was determined by Western-blotting in total protein extracts. In some experiments, the EPs® 7630 pre-treated cells were infected with 1 MOI RV-16 for up to 48 hours. Total proteins were collected over 4 days and analysed as described below for protein expression, or cells grown on cover slips were fixed with 2% formaldehyde for 2 x 5 min. followed by staining and fluorescence microscopy for RV16 protein expression (Fig. 1b).
Rhinovirus infection and detection: The RV-16 strain used was described earlier (Oliver et al 2008). Cells were seeded in 8-well chamber slides (Thermo Fisher Scientific, Switzerland), and at 80–90% confluence they were infected with RV16 (1x multiplicity of infection: MOI) by 5 min centrifugation (200 x g). Cells were continued under standard cell culture conditions for up to 4 days. RV16 infection was monitored by immunofluorescence for anti-RV16 antibody (cat# 18758, QED-Bioscience Inc. San Diego, USA). Cells were fixed by formalin (4% in PBS, 2 x 5 minutes), washed twice (PBS), and permeabilised (5 min., 0.01% TWEEN-100 in PBS). Unspecific antibody binding was blocked (30 min., 2% bovine serum albumin in PBS) and afterwards incubated with the anti-RV16 antibody (1:100 dilution, overnight, (4oC). Cells were washed 3 x (PBS), followed by incubation with anti-mouse FITC labelled antibody (Abcam, Switzerland, 1 hour, room temperature). Nuclei were stained by DAPI for cell counting (Thermo Fisher Scientific). The number of RV16 positive cells was determined after 3 washes (PBS) by immunofluorescence microscopy (EVOS FLoid cell imaging station, Thermo Fisher Scientific). All experiments were performed in a Bio-Safety-Level-II laboratory.
Cytosolic - nuclear protein translocation: Confluent epithelial cells were treated with EPs® 7630 (10 µg/ml) for 48 hours before being stimulated with increasing concentrations of vitamin D (0.1–10 mM) over various time periods (0, 3, 6, and 24 hours). The cell compartmental distribution of the VDR was determined by immunofluorescence staining using EVOS cell imaging system (Thermo Fisher Scientific, Waltham, USA).
Western-blotting: Cells were lysed in RIPA buffer, or as cytosolic and nuclear proteins. The protein content was quantified by BCA (Thermo Fisher Scientific). Denatured proteins (10µg) were size-fractionated by electrophoresis (8–16% SDS–PAGE, Thermo Fisher Scientific), and transferred onto PVDF membranes. Unspecific binding of antibodies was blocked by 30 min incubation of the membranes with 2%bovine serum albumin in phosphate buffer saline (PBS) containing 0.05% TWEEN-20. Proteins were detected by incubating the membranes with one of the primary antibodies to either the VDR, Erk1/2 MAPK, p38 (α, β, γ, δ), JNK, E-cadherin, or GAPDH (all: Abcam Plc, Cambridge, UK) for overnight at 4oC. Following 3 washes with blocking buffer, the membranes were incubated with secondary species-specific HRP conjugated antibodies (Abcam). Protein bands were visualised by chemiluminescence, applying SuperSignal West Dura substrate (Thermo Fisher Scientific) and documented by c300 (Azure Biosystems, Dublin, USA).
Immunofluorescence: Epithelial cell were seeded on 8-well PCA-slides (cat 94.6140.802, Sarstedt, Sevelen, Switzerland) and allowed to adhere overnight. Cells were then treated with medium alone, or by increasing concentrations of EPs® 7630 (0.01-10 µg/ml) over various durations up to 5 days. Cells were fixed in 4% paraformaldehyde (in PBS, 2×5 min), and immuno-fluorescence staining was performed as described earlier [7]. Nuclei were stained by DAPI.
Statistics: The Null-hypothesis was: No modification of protein expression, activation or location by any treatment compared to untreated cells. The Null-hypothesis was tested by ANOVA, Student’s t-test and sub-sequent Mann-Whitney U-test as appropriate; p-value < 0.05 was accepted as significant.
{kind=link}