Benchmarking Results. In this work, we assess the accuracy and efficiency of the introduced LaDyBUGS method compared to experiment and a community standard alchemical free energy method (TI/MBAR). Free energies of binding were calculated for 24 ligands bound to one of three benchmark protein systems: DNA ligase, MUP1, or c-Met. Figure 5 plots the correlation between experiment and computed ΔGbind with LaDyBUGS (15 ns) and TI/MBAR (5 ns/window); all data points are reported in Table S1 of the Supplementary Information. Root mean square error (RMSE) and Kendall τ scores85 were computed for each test system individually and for the combined dataset. Using these metrics, we see a uniform improvement in both RMSE and Kendall τ with LaDyBUGS relative to TI/MBAR. For all 24 ligands, the LaDyBUGS RMSE was 0.89 kcal/mol and the Kendall τ was 0.67. For every test case, the calculated LaDyBUGS RMSE was about or below 1.0 kcal/mol, a typical goal and state-of-the-art for predictive accuracy in free energy calculations for drug discovery.5–7, 11,16,17,24 It is important to note that accuracy is dependent on both correct force field representation of a chemical system and thorough configurational sampling with a given free energy method.86 The larger RMSE of 1.03 kcal/mol and reduced Kendall τ of 0.59 from TI/MBAR (5 ns/window), which used the same force field parameters as LaDyBUGS, suggests LaDyBUGS is providing improved sampling proficiency over TI/MBAR for the same benchmark systems. This seems extraordinary, since LaDyBUGS used 19.2 times less sampling than TI/MBAR (5 ns/window) (Fig. 6).
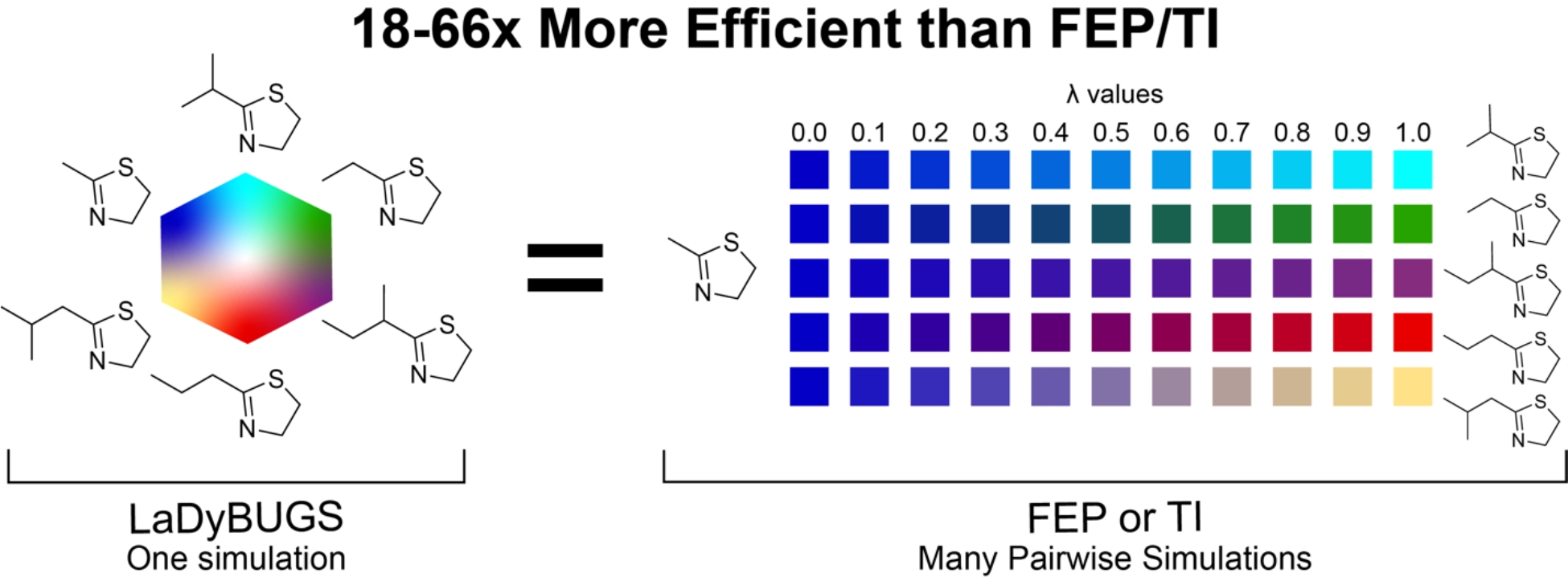
Because both LaDyBUGS and TI/MBAR calculations used the same force field parameters, we can also compare the precision of their ΔGbind predictions (Fig. 6). The two computational methods agree well with each other, with an overall RMSE of 0.43 kcal/mol. Considering that most protein-ligand ΔGbind calculations have computed uncertainties between 0.3–0.5 kcal/mol,16,48,49 and that LaDyBUGS bootstrapped errors ranged from 0.08–0.48 kcal/mol (Table S1), these results suggest good agreement exists between these free energy methods. To explore the effect of sampling time on the ΔGbind results, we also compared these methods with less sampling per LaDyBUGS (5 ns) simulation and more sampling per TI/MBAR calculation (15 ns/window). In Fig. 7, agreement between LaDyBUGS simulations with only 5 ns of sampling compared to TI/MBAR (5 ns/window) remains high with a RMSE of 0.40 kcal/mol and a Kendall τ of 0.83. As expected with a reduction in sampling, the mean bootstrapping error for LaDyBUGS increased from 0.21 to 0.37 kcal/mol, although the RMSE between LaDyBUGS (5 ns) and experiment remains about the same at 0.92 kcal/mol. This level of agreement seems remarkable considering LaDyBUGS (5 ns) used 57.8 times less sampling on average than TI/MBAR (5 ns/window). For the DNA ligase test case, LaDyBUGS used 66 times perturbations in all benchmark systems, for the protein-bound edge of the thermodynamic cycle less sampling than TI/MBAR. Thus, from a total of 60 ns expended to sample 24 alchemical (Fig. 1), LaDyBUGS (5 ns) provided ΔGbind predictions with errors at or below 1.0 kcal/mol compared to experiment. On a per-ligand basis, LaDyBUGS spent only an average of 2.5 ns of protein-ligand sampling. These results highlight that significant cost savings are achievable with LaDyBUGS without compromising accuracy in the computed ΔGbind results. In contrast, TI/MBAR required almost 3.5 µs of total protein-ligand sampling (165 ns/ligand), without employing commonly used redundant calculations for cycle closure and hysteresis error reduction.18–20 To test LaDyBUGS convergence, 25 ns simulations were also run. No large deviations were observed and the RMSE between 15 ns LaDyBUGS and 25 ns LaDyBUGS simulations was 0.11 kcal/mol, well within statistical noise (Table S1). The RMSE of 0.23 kcal/mol between 5 ns and 25 ns LaDyBUGS results was slightly larger but still within noise, suggesting a high degree of convergence even with a minimal amount of LaDyBUGS sampling. Figure 7 also shows the effects of extending TI/MBAR sampling to 15 ns/window for the DNA ligase and c-Met systems. Large improvements are observed in comparison to TI/MBAR (5 ns/window). The RMSE to experiment improves to 0.93 kcal/mol, and the RMSE to LaDyBUGS decreases to 0.27 kcal/mol. The strong agreement between short 5 ns runs of TI/MBAR and LaDyBUGS, as well as between the longer 15 ns runs of TI/MBAR and LaDyBUGS, suggests LaDyBUGS is able of deliver comparable accuracy as TI/MBAR with significant cost savings in terms of sampling (18–66 times less simulation time required). Improved efficiency with LaDyBUGS directly stems from its ability to investigate several alchemical perturbations collectively within a single simulation, without the need to break up transformations into separate λ windows spread across multiple separate simulations (Fig. 2).
Uniformity of λ Sampling with LaDyBUGS. One potential issue associated with expanded ensemble free energy methods is a difficulty in achieving sampling smoothness of all λ states, e.g., avoiding becoming stuck primarily sampling one or several states too often and neglecting to sample all other λ states.41,87 This problem has been observed in an expanded ensemble investigation that used a Wang-Landau algorithm to propagate λ switching.28 In conventional λD simulations, including d-GSλD, static biases are added to a simulation to reduce free energy barriers between λ states and facilitate transitions between λ states.41,47 In most situations these biases work well, all λ states are evenly sampled, and reliable free energy predictions are obtained. But burn-in time is required to first identify appropriate biases for these methods, which decreases their overall efficiency. Furthermore, if the protein-ligand system experiences a rare event during production sampling, such as a conformational change associated with a slow degree of freedom, static biases may be mismatched to the free energy surface of the new conformation and thus unable to facilitate continuous λ sampling; the simulation would become trapped, requiring new biases to be identified and sampling to be restarted. However, the dynamic biases used in LaDyBUGS continuously propagate the sampling of many λ states without prior burn-in time for bias identification and allow for conformational plasticity of the chemical system without getting trapped in a small number of λ states. Biases from Eq. 4 rely solely on the number of times each λ state has been sampled and on-the-fly FastMBAR free energy estimates; thus, LaDyBUGS can provide incredibly smooth λ sampling throughout an entire simulation. Figure 8 shows the difference between the minimum and maximum number of times a λ state was sampled as a function of time, referred to as “counts”, averaged across all protein simulations used for benchmarking. The difference does not exceed a count of 4, even though λ states are sampled more than 500–800 times by the end of each simulation. This level of sampling smoothness ensures that LaDyBUGS does not become trapped in λ sampling and provides rapid transitions between multiple ligand end states for accurate free energy calculation with FastMBAR.
LaDyBUGS Samples a Mixture Distribution of λ States. Smooth transitions between states are also facilitated by strong energetic overlap between neighboring λ states. In our benchmark studies, c-Met group 1 consists of different 5-membered heterocycles while group 2 contains a mixture of carbamate and aryl substituents (Fig. 4). As shown in Fig. 9 for two example c-Met group 1 and 2 perturbations, a uniform Δλ schedule provides good energetic overlap between both similar (c-Met Group 1) and dissimilar (c-Met Group 2) transformations. This enables facile transitions to adjacent λ states when sampling the P(λ|X) conditional distribution (Fig. 3). As shown in Fig. 9, most transitions occur to + 1 or + 2 states away, although large jumps (> 4 states) are sometimes observed. The degree of overlap between λ states affects the transition distance traveled, with higher overlap facilitating larger jumps (see also Table S2). The mean transition distance traveled for c-Met group 1 is 2.47 states, but it is smaller at 1.64 states for c-Met group 2 which has less overlap between adjacent states (Fig. 9). Fortuitously, transitions between energetically similar and adjacent λ states enables the chemical system to quickly relax and equilibrate during the brief 200 ps MD simulation whenever a new λ state is sampled. Therefore, we assume the MD configuration drawn from P(X|λ) in each GS step represents an equilibrium sample. By constant sampling of different λ states and atomic coordinates, LaDyBUGS can efficiently sample a mixture distribution of λ states within a single simulation. Pairing free energy determination with the MBAR algorithm is natural then, because MBAR pools and reweights samples as if they originated from a mixture distribution.15,88 Our proof in the SI demonstrates that samples drawn from the same λ state with different external biases can be treated as coming from the same state. We then use FastMBAR to obtain equilibrium free energy results from a LaDyBUGS simulation, under the stated assumptions of the proof. Furthermore, because sampling of the P(λ|X) conditional distribution requires energies to be calculated for every λ state at every sampled P(X|λ) configuration, no postprocessing of LaDyBUGS trajectories is required to run MBAR; all necessary information is generated on-the-fly and available at the conclusion of a LaDyBUGS simulation.
Software Implementation. LaDyBUGS has been implemented in OpenMM51 and all LaDyBUGS scripts are available for download on the Vilseck-Lab GitHub page. One advantage of using OpenMM for LaDyBUGS is the ability to use force groups to partition the interactions of different components of an alchemical system and thus enable λ state-dependent energies to be evaluated without recalculating the energy of the entire chemical system. This feature speeds up the sampling of P(λ|X) which requires λ-dependent energies to be calculated for every λ-state at every P(X|λ) configuration. Consequently, we find that sampling a group of 6 ligands collectively with 141 λ states is only marginally slower than performing a standard pairwise perturbation of 11 λ states with LaDyBUGS. For example, on a NVIDIA 2080 TI graphics processing unit (GPU), 6 duplicate 5 ns LaDyBUGS c-Met group 1 cmet_9 → cmet_10 pairwise perturbations each took ca. 10.85 hours to run. Similarly, 6 duplicate 5 ns simulations of all 6 c-Met group1 ligands sampled collectively took ca. 11.01 hours each. Thus, the combined 6-ligand calculation was only ~ 1.5% slower, highlighting the effectiveness and cost-savings of sampling multiple ligands simultaneously with LaDyBUGS. With our current implementation of LaDyBUGS in OpenMM and using an assumption of sampling 6 perturbations per LaDyBUGS simulation, we estimate that ca. 4–13 compound perturbations can be investigated per day per 1 GPU with LaDyBUGS using a range of 15 ns to 5 ns of sampling per calculation, respectively. On a modest cluster of 25 GPUs, this readily scales to 100–325 perturbations per day! Hence, rapid high-throughput screening of hundreds of lead compound analogues with highly accurate free energy predictions is obtainable with LaDyBUGS within a day using minimal computational resources.
Work is on-going to further optimize our implementation of LaDyBUGS in OpenMM as well as incorporate it into other software suites, including CHARMM. If a program lacks the ability to partition energetic interactions via a “force group”-like algorithm, P(λ|X) may be sampled by calculating the energy of the entire chemical system; all non-alchemical environment-to-environment interactions should cancel out when λ state-dependent energies are compared. Though some wall-time slowdown may be expected to occur as a consequence of running a larger energy evaluation, we anticipate that LaDyBUGS would still provide highly efficient results, nonetheless. Incorporating LaDyBUGS into CHARMM, or other programs, could provide additional benefits too. For example, the CustomNonbondedForce class in OpenMM makes it challenging to use particle mesh Ewald (PME) methods with LaDyBUGS. However, a λD-based PME approach is already available in CHARMM and BLaDE for running MSλD simulations,45,76,77,89 and this could be utilized with LaDyBUGS in CHARMM to facilitate the inclusion of long-range electrostatic interactions in future calculations.
Multisite sampling. Finally, we emphasize that the efficiency gains for LaDyBUGS reported in this work used only single site perturbations, where substituent group modifications occurred at only one site off a central ligand core. Multisite perturbations, with functional group substitutions occurring at multiple sites around a ligand core, could also be accomplished by using the previously described d-GSλD approach for creating two- or three-dimensional λ states.47 Such LaDyBUGS simulations may need longer total sampling to obtain converged results due to the increased number of λ states required for multisite sampling, but this has not been tested yet. Instead, this work focused on single site perturbations to match structure-activity relationship strategies typically pursued experimentally by changing one component of a lead compound at a time.16,56−58 In this manner, LaDyBUGS seems especially adept at exploring incremental changes to a lead compound. Future investigations will reveal the applicability of LaDyBUGS to tackling larger or more challenging perturbations or for molecular decoupling to compute absolute free energies of binding directly.
{kind=link}