In-silico simulations of protein preparation, ligand preparation, grid generation, molecular docking, and molecular mechanics were carried out in the Maestro11.9.011modeling package provided by Schrödinger, LLC, New York, NY, 2019-1, installed in a Dell OptiPlex 3060 with a processor Intel Core i7-8700, Ubuntu 16.04 LTS, graphics GeForce GT 730/PCle/SSE2, and a 64-bit operating system Energetically optimised structure-based pharmacophore generation and molecular dynamics were carried out in the Maestro12.4modeling package provided by Schrödinger, LLC, New York, NY, 2020-2, installed in a Dell Precision 7820 with an Intel Xeon(R) Gold 6130 processor, Kernel GNOME Version 3.28.2CentOS Linux 7, Graphics Quadro P5000/PCle/SSE2, and 64-bit OS.
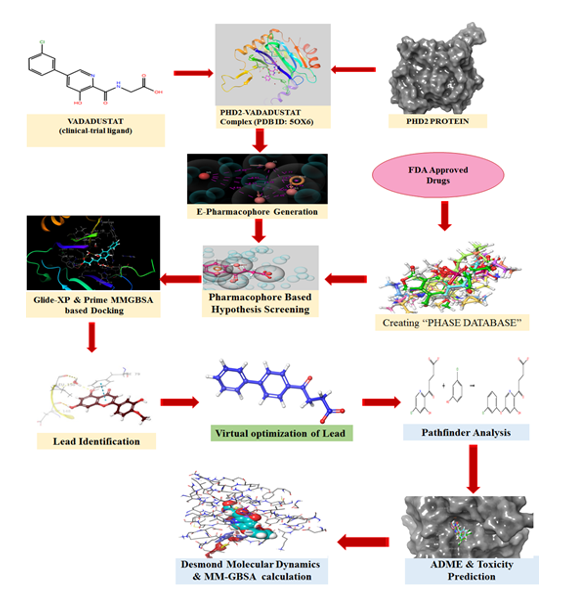
The Energetically optimized structure-based pharmacophore (e-pharmacophore), is both a ligand and structure-based approach to obtain structurally diverse active ligands from FDA approved drug dataset. The 3-Dimensional crystal structure of human PHD2 protein (PDB ID: 5OX6) co-crystalized with potent inhibitor (Vadadustat) was obtained from the Protein Data Bank (PDB). Simultaneously, the list of FDA approved drug candidates was collected from Drugbank database. Initially, the obtained raw protein (5OX6) was pre-processed and energetically minimized with the assistance of the OPLS3e force field to get a refined protein structure using the “Protein Preparation Wizard” panel of GLIDEv7.7 Module provided by Schrodinger suite (Schrödinger, LLC, New York, NY, 2019-1).
In addition, the reference inhibitor Vadadustat was extracted and produced from the co-crystallized protein using GLIDEv7.7's "Ligprep" settings (Schrödinger, LLC, New York, NY, 2019-1). The generated ligands were then re-docked in Extra precision mode (XP-docking algorithm) with previously prepared PHD2 protein and the top-ranking docked posture was recovered for further hypothesis creation. The docking methodology was confirmed by computing the RMSD values by superimposing reference ligand (Vadadustat) re-docked conformations with the original co-crystallized conformation of a ligand. The reward and penalty areas of the reference ligand were visualised using the XP visualizer programme in the GLIDEv7.7 module (Schrödinger, LLC, New York, NY, 2019-1).
The fundamental chemical properties necessary for the creation of e-pharmacophore hypotheses were chosen based on the Glide XP descriptor information. Using the PHASE v3.8 module of the Schrodinger suite (Schrödinger, LLC, New York, NY, 2020-2), a customized e-pharmacophore hypothesis including five critical chemical characteristics (AAARN) that enhance ligand binding was constructed using accessible protein-ligand complexes. The PHASE v3.8 module was used to generate a phase database for database screening of the previously stated FDA medications (Schrödinger, LLC, New York, NY, 2019-1). Using the phase database, the derived pharmacophore hypothesis was efficiently evaluated for shape-based similarity. This hypothesis screening allows the selection of ligands from the phase database comprising a list of FDA authorised medications that have comparable chemical properties to the reference ligand.
To identify the potentially interacting drug candidates from the database, the compounds obtained after database screening were docked with the active pocket of the minimized protein (5OX6) using the same XP docking mode (GLIDEv7.7) as that of reference ligand. The compounds which possessed better docking scores and protein interaction were selected for further analysis of binding free energy calculation, pharmacokinetic & toxicity profile, and protein-ligand stability predictions. MM-GBSA tool in the Prime v3.5 module(Maestro11.9.011, Schrödinger, LLC, New York, NY, 2019-1) was utilized to calculate the binding free energy of selected ligands[11].
Experimentally more relevant absorption, distribution, metabolism, and excretion properties of selected drug candidates were predicted theoretically using QikProp v5.4, a more precise rapid software package incorporated in the Schrodinger suite (Maestro11.9.011, Schrödinger, LLC, New York, NY, 2019-1). Additionally, Qikprop was used to check Lipinski's rule-of-five and Jorgensen's rule-of-three infringements to determine whether a compound is drug-like [12].
MOLECULAR DYNAMICS
The ligands scrutinized from the above interpretations were subjected to analyze the stability, RMSD, RMSF, and interaction from the protein-ligand complex in a more relevant physiological environment by running MD simulations using the OPLS_2005 force field in DESMOND V5.2 module of Schrodinger suite (Schrödinger, LLC, New York, NY, 2020-2)[13]. Firstly, the system builder was constructed by placing the protein-ligand complex inside the orthorhombic boundary box containing TIP3P water as a solvent model. The protein was again minimized and recalculated to add more suitable counterions to neutralize the charges of the system. Additionally, 0.15 M NaCl salt concentration was fixed in the system ,20Aº away from the ligand to simulate background salt at the physiological condition. The above-prepared system was incorporated in the workspace to start an MD simulation. Molecular dynamics were run with NPT ensemble at 300K temperature and 1.01325 bar atmospheric pressure for 100ns using the OPLS_2005 force field. The Structure frames computed for every 50 ps were saved in trajectory during molecular dynamics; a total of 1000 frames were saved. The generated MD results were further analyzed through the simulation interaction analysis tool in the DESMOND V5.2 module[14-15].
{kind=link}