Alzheimer's disease (AD) is a neurodegenerative age related brain disease which diminishes learning and memory. Two common cholinesterases (ChEs), first acetylcholinesterase (AChE) and second one butyrylcholinesterase (BChE) are the main targets to treat this disease. In the current paper, six novel pyrazolo-benzothiazole analogs were systematically planned, synthesized, and biological tested for their ChEs activities, antioxidant, enzyme kinetic, self induced Aβ modulation, PAMPA-BBB, and docking and molecular simulation study. Among all derivatives,4a bearing 3,4-dimethoxypyrazolo-benzothiazole molecules was the potential inhibitor of AChE (IC50 = 8.69 ± 0.42 µM), Aβ modulator and good antioxidant. Docking studies also clearly revealed that compound 4a fit into the active sites of hAChE.
Research Article
Design, Synthesis, and biological evaluation of pyrazolo-benzothiazole derivatives as a potential therapeutic agent for the treatment of Alzheimer’s disease
https://doi.org/10.21203/rs.3.rs-1624677/v1
This work is licensed under a CC BY 4.0 License
You are reading this latest preprint version
Alzheimer’s disease (AD)
chalcone
pyrazole
acetylcholinesterase
Alzheimer's disease (AD) is a multifactorial, age-related neurological brain illness caused by the loss of cholinergic neurons and synapses in the cerebral cortex, cortical and subcortical brain areas. [1] In the year 2020, there will be more than 55 million people living with dementia all over the world. Every 20 years, the population will nearly double, to 78 million in 2030 and 139 million in 2050. Although the exact pathophysiology of AD is unknown, literature suggests that low levels of the neurotransmitter acetylcholine (ACh), amyloid beta (Aβ) plaque deposition, hyperphosphorylation of tau protein in the form of neurofibrillary tangles, increased oxidative stress, and metal dyshomeostasis all play a role in the progression of AD. [2, 3] Acetylcholine (ACh) is a cholinergic neurotransmitter that plays a role in memory and learning. [4] Cholinergic pathways in the CNS are disrupted in Alzheimer's disease, and the resulting cholinergic deficit adds to cognitive impairment. [5] Furthermore, a decrease in cholinergic activity in the CNS of AD patients is linked to a worsening of the dementia scale. [6] There is currently no permanent cure for Alzheimer's disease, and FDA-approved medications for the treatment of the disease are only intended to provide symptomatic relief. [7] Approved medications only provide symptomatic relief or decrease the worsening of symptoms when used alone or in combination, but they do not halt or stop the progression of the disease. [8] The pharmacological agents approved for AD management have some beneficial effects on cognitive, and behavioral symptoms of the disease, however, these agents do not play any role to address basic aspects of the disease.[9] Moreover, these agents are neuro-destructive rather than having neuroprotective effect.
There is practically no medicine for the treatment of Alzheimer's disease that can address the illness's basic pathophysiological components. [10] Acetylcholinesterase (AChE) inhibitors and N-Methyl-D-aspartic acid or N-Methyl-D-aspartate (NMDA) receptor antagonists are currently available for the treatments of Alzheimer's disease. Donepezil (1), rivastigmine (2), galantamine (3), and tacrine (4, taken from the market) are AChE inhibitors, while memantine (5) is an NMDA receptor antagonist (Figure 1). [11] AChE is a serine protease enzyme that hydrolyzes adenosine triphosphate (ACh) into choline and acetic acid. [12] As a result, it is extremely important in cholinergic neurotransmission. The catalytic anionic site (CAS), the oxyanion hole, and the peripheral anionic site are the three parts of AChE's binding site (PAS). [13] Next, butyrylcholinesterase (BChE), an isomer of ChE that is responsible for the cleavage of ACh, is found in the brain. [14] As the disease advances, the levels of both AChE and BChE fluctuate dramatically. [15] As a result, both enzymes have been investigated as potential neuroprotective and disease-modifying therapies for Alzheimer's disease. In the last two decades, research on Alzheimer's disease has yielded new insights into core pathogenic processes such as the accumulation of misfolded proteins A in aggregates, oxidative stress, and elevated metal concentrations in the AD brain. [16] The generation of reactive oxygen species can be caused by oxidative stress and an excess of metals (ROS). [17] These ROS impair mitochondrial respiration and promote the formation of intracellular neurofibrillary plaques and tangles from amyloid-beta (Aβ) plaques.
Next, butyrylcholinesterase (BChE), isomer of ChE, is also present in the brain responsible for cleavage of ACh.[14] The level of both AChE and BChE changes intensely as the disease progresses.[15] Therefore, both enzymes have been explored for the neuroprotective and disease modifying therapy for AD. The research in AD in the past two decades has provided newer insights into the basic pathogenic factors such as accumulation of misfolded proteins Aβ in the form of aggregates, oxidative stress, and increased concentration of metals in the AD brain.[16] Oxidative stress and excessive amounts of metals can lead to the formation of reactive oxygen species (ROS).[17] These ROS inhibits mitochondrial respiration and promote the aggregation of amyloid-β (Aβ) plaques in the form of intracellular neurofibrillary plaques and tangles.
Chalcones (6) are naturally occurring molecules and their synthetic compounds display a variety of biological activities and are precursors of natural flavonoids derivatives which are present in vegetables, fruits and various medicinal plants (Figure 2).[18] Natural flavonoids contain OCH3 and OH groups at different positions on aromatic rings. Both the aromatic rings are linked through a three-carbon α, β-unsaturated carbonyl group with structural diversity that can be achieved by naturally (eg. broussochalcone A, xanthohumol and dixanthohumol) and synthetically.[19] They also serve as an important precursor for the synthesis of varieties of heterocyclic systems. Chalcones exhibit wide class of pharmacological activity related to the neurodegenerative disorder including anti-inflammatory, antioxidant, cholinesterase inhibition, and Aβ modulation properties.[20] These finding clearly suggest that chalcone could be used as a carrier linker in the development of new class of multifunctional compounds for the management of AD.
Pyrazole scaffold enormously plays a significant role in medicines and many functional derivatives are commercially available.[21] Due to their distinct synthetic and medicinal utility pyrazole remains to be robustly explored in new drug design and development. Pyrazoles are readily able to show interactions with numerous enzymes located on the target cells in biological system including cyclooxygenase, α-glycosidase, carbonic anhydrase and acetylcholinesterase.[22]
Pyrazole analogues served as an essential core scaffold in many pharmaceutical drugs and agrochemicals namely mepiprazole, celecoxib, rimonabant, phenazone, ruxolitinib, deracoxib etc. A series of studies revealed that numerous modulated analogues have been reported in the literature regarding the pharmacological activities of pyrazoles.[22-24] Some of these analogues depicted potent biological activities such as, analgesic, anti-inflammatory, antipyretics, and treating erectile dysfunction etc. New pyrazole-containing lead molecules that exhibits favorable ADME/therapeutic index, improved potency, target specificity which might be considered as an area of interest for prospective researchers working on this heterocycle.[25]
Therefore, a series of pyrazole analogs were systematically designed based on multifunctional property. Developed compounds were further biological evaluated for their inhibition of ChEs, enzyme kinetic, antioxidant, Aβ modulation and in-vitro BBB permeability study. Molecular docking and dynamic studies were also dipicted in order to get the detailed into the binding interaction of the developed molecules with enzyme.
2.1. Chemistry
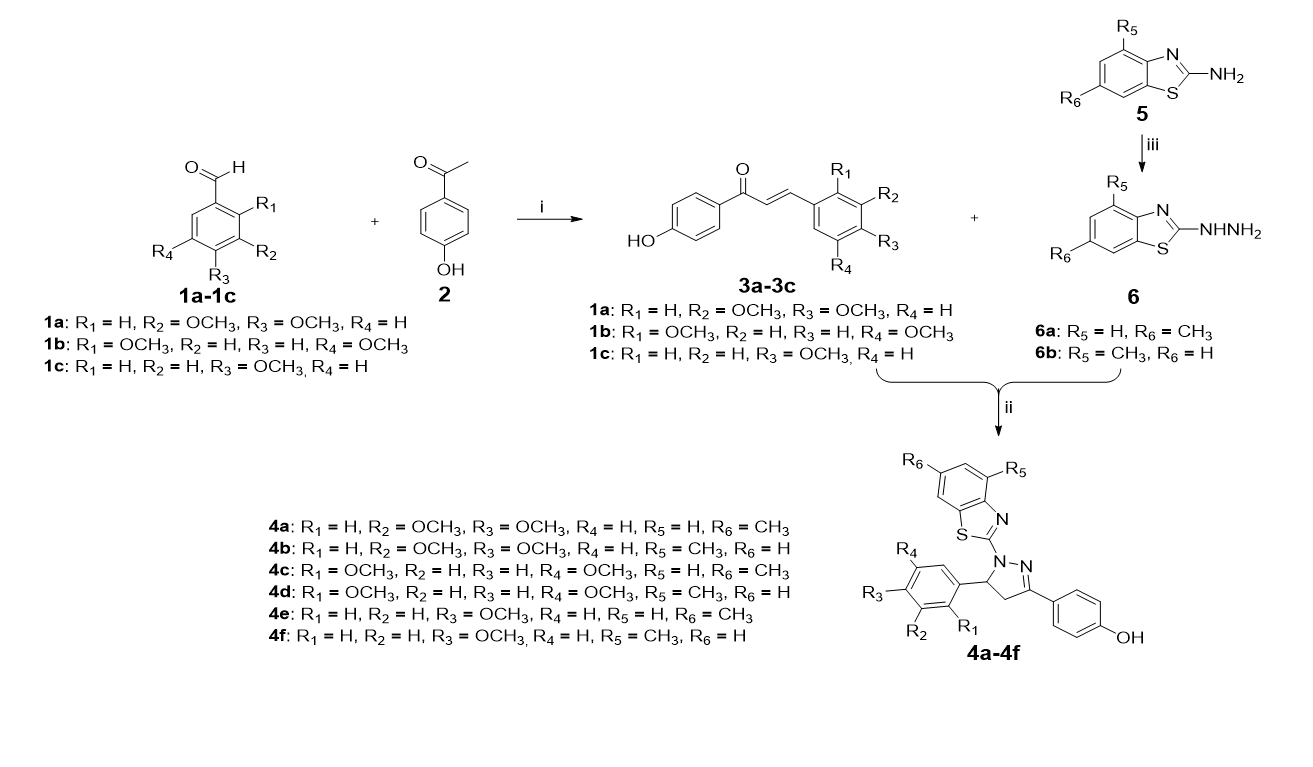
The complete synthetic route of pyrazolo-benzothiazole analogs were presented in scheme 1. In first step, 4-hydroxyactophenone was treated with different mono/disubstituted aldehyde in the presence of NaOH and solvent ethanol to afford substituted chalcone as a key intermediate 3a-3c. The 1H-NMR spectra of 3a-3c showed characteristic two doublet (d) peak for E-alkene of chalcone at 7.66-7.80 ppm with coupling constant (J) 15.50 Hz clearly indicate E-configuration for developed intermediates. While second intermediate were prepared by the reaction of substituted 2-aminobenzothiazole with hydrazine hydrate, ultimately lead to the formation of substituted hydrazine derivative (6a-6b). Finally, the desired compounds 4a-4f were obtained by the simple cyclization of intermediate 3a-3c with substituted hydrazino-benzothiazole (6a-6b) in n-butanol at 120 0C.These final compounds have not been previously mentioned in the literature. Their structures analysis were analyzed by 1H NMR, and 13C NMR spectroscopy.
2.2. Rationale and Design
We started our drug design approach by selecting the chalcone scaffold as a starting point. The chalcone consist of two parts (i) acetophenone fragment and (ii) aldehyde fragment, therefore, impart antioxidant potential to chalcone. Furthermore, benzothiazole is an important pharmacophore for imparting Aβ modulation property. Therefore, the concept of hybridization was used for the development of novel pyrazolo-benzothiazole derivatives, which is based on the connection of two fragments (chalcone and hydrazino-benzothiazole) via a suitable dihydropyrazole fragment (Figure 3).
2.3. Biological activity
2.3.1 Evaluation of AChE and BChE inhibitory activities
The inhibitory activities of the discovered compounds 3a-3f and 4a-4f against hAChE (human serum) and eqBChE (equine serum) were determined using a modified Ellman colorimetry assay. A well-known medication, donepezil (DNP), was employed as the control. The inhibitory actions of the synthesised analogues against AChE and BChE are listed in Table 1 as percent inhibition and IC50 values. When compared to their chalcone intermediates, all of the synthesized analogues displayed good to low ChE inhibitory activity against AChE and BChE. (3a-3f). Almost all of the resulting analogues had significant selectivity for hAChE over eqBChE and showed good inhibitory potency in AChE (IC50 ranging from 8.69 0.42 to 18.55 0.51 μM).
2.3.2. SAR Analysis
To validate our drug design approach, firstly chalcone analogs (3a-3c) was syntheiszed and tested for their cholinesterase inhibitory (ChEI) activity. To our interest, as mentioned on Table 1. Chalcone analogs (3a-3c) presented % AChE inhibition (at 20 μM) from 11.36-31.14%. This result of the chalcone scaffolds encouraged us to further explore this template. Then, we cyclize chalcone system with hydrazino-benzothiazole to generate compounds 4a-4f. All the developed compounds showed potent ChEs inhibitory property compared to the chalcone analogs (3a-3c). Among all compound 4a presented highest ChE inhibitory potency with IC50 value of 8.69 ± 0.42 μM against AChE, and 15.36 ± 0.33 μM against BChE. SAR study revealed that mono substituted (-OCH3) pyrazolo-benzothiazole (3e and 3f) showed poorer ChEs activity compared to the disubstituted(-OCH3) pyrazolo-benzothiazole derivatives (IC50, AChE (μM); 8.69 ± 0.42 for 4a, 11.23 ± 0.61 for 4b, 17.23 ± 0.39 for 4c, 15.27 ± 0.45 for 4d, 14.62 ± 0.36 for 4e, and 18.55 ± 0.51 for 4f). While in case of BChE all the developed compounds showed mixed activity IC50 value from 15.36 ± 0.33 to 19.21 ± 0.44 μM. Furthermore, some compounds such as 4d, 4e, and 4f represented poor BChE inhibition with % inhibition of 24.63 ± 0.71, 27.25 ± 0.48, and 34.81 ± 0.66, respectively at 20 μM concentration. It has been observed that introducing additional methoxy (-OCH3) to the benzene ring increases ChE inhibitory activity compared to monosubstituted one (Supporting figure S1).
Table 1
ChE inhibitory activities, antioxidant, and PAMPA-BBB permeability by 3a-3f, and 4a-4f, and donepezil (DNP).
|
Compd Code |
hAChE IC50 (µM)a/ % inhibitionb |
eqBChE IC50 (µM)a/ % inhibitionb |
DPPH IC50 (µM)/% inhibitiona |
Pe (106 cm s-1) |
Permeability prediction (CNS+/-) |
Melting point (oC) |
|
3a |
31.14 ± 0.53% |
19.26 ± 0.78% |
87.32 ± 1.23 |
ndc |
ndc |
140-145 |
|
3b |
24.36 ± 0.66% |
15.21 ± 0.86% |
97.63 ± 1.31 |
ndc |
ndc |
136-140 |
|
3c |
20.56 ± 0.41% |
11.63 ± 0.72% |
105.04 ± 1.02 |
ndc |
ndc |
132-135 |
|
4a |
8.69 ± 0.42 |
15.36 ± 0.33 |
59.65 ± 1.43 |
8.36 ± 0.56 |
CNS (+) |
170-172 |
|
4b |
11.23 ± 0.61 |
18.23 ± 0.28 |
66.35 ± 1.32 |
7.21 ± 0.41 |
CNS (+) |
166-170 |
|
4c |
17.23 ± 0.39 |
19.21 ± 0.44 |
71.13 ± 1.11 |
6.89 ± 0.61 |
CNS (+) |
157-160 |
|
4d |
15.27 ± 0.45 |
24.63 ± 0.71% |
75.25 ± 1.19 |
6.45 ± 0.36 |
CNS (+) |
160-165 |
|
4e |
14.62 ± 0.36 |
27.25 ± 0.48% |
88.63 ± 1.32 |
4.55 ± 0.42 |
CNS (+) |
172-175 |
|
4f |
18.55 ± 0.51 |
34.81 ± 0.66% |
91.51 ± 1.09 |
5.32 ± 0.66 |
CNS (+) |
170-175 |
|
DNPe |
0.96 ± 0.31 |
2.36 ± 0.61 |
nad |
14.53 ± 0.54 |
CNS (+) |
na |
|
TPf |
nad |
nad |
27.32 ± 0.62 |
nad |
nad |
na |
|
ATNg |
nad |
nad |
nad |
1.36 ± 0.63 |
CNS (-) |
na |
|
DZ |
nad |
nad |
nad |
15.63 ± 0.56 |
CNS (+) |
na |
aIC50 values represent the concentration of inhibitor required to decrease activity by 50% and are the mean of three independent experiments, each performed intriplicate (SD = standard deviation).b Percentage inhibition at 20 µM concentration of inhibitor. cnd = Not determined. hAChE from human serum. dna = not applicable. eqBuChE from equine serum. eDNP = Donepezil. fTP = Tocopherol. gATN = Atenolol. hDZ = Diazepam
2.3.3. AChE inhibition kinetic studies
In order to investigate the method of enzyme inhibition, enzyme kinetic study was performed with the most potent inhibitor of AChE (i.e., 4a) against hAChE. The categories of inhibition was examined by a Lineweaver-Burk double reciprocal plot with the velocity of the substrate acetylthiocholine iodide (y-axis) and increasing concentrations. The plots (Figure 4) clearly indicate mixed inhibition pattern of 4a against hAChE. From this study we can conclude that 4a binds to both free AChE and AChE substrate complex.
2.3.4. Evaluation of antioxidant activity
It is apparent from the literature that oxidative stress plays a significant role in the progression of AD [26]. Drugs inhibiting the generation or clearing of the free radicals in the brain would be advantageous for AD treatment. Therefore, the antioxidant activities of the developed molecules were evaluated using DPPH assay. DPPH radical can be used in initial testing of analogs capable of scavenging radical species. For comparison, tocopherol (TP) was used as reference. All the developed derivatives (4a-4f) presented potent radical scavenging activity compared to the intermediate analogues (3a-3c). All the synthesized compounds showed IC50 value from 59.65 ± 1.43 to 91.51 ± 1.09 μM. Among the developed molecules compound 4a exhibited highest DPPH radical scavenging capacity with IC50 = 59.65 ± 1.43 μM. This study clearly indicates that developed compounds represent potential antioxidant activity (Supporting figure S2).
2.3.5. Inhibition of the self-mediated Aβ1-42 aggregation
The ChEs inhibitor which bind to PAS region are competent of inhibiting Aβ1-42 aggregation. In order to estimate Aβ modulation property of compound 4a, thioflavin T (ThT) fluorescence experiment was performed. To perform this experiment, Aβ alone, Aβ + DNP (10:10 µM), Aβ + DNP (10:20 µM), Aβ + 4a (10:10 µM), and Aβ + 4a (10:20 µM) was incubated for 48 hr at 37 oC. The results were presented as normalized fluorescence intensity (NFI) (Figure 5). Compound 4a, at a concentration ratio of 10:10 µM and 10:20 µM, exhibited potent Aβ modulation property compared to self-induced Aβ aggregation. These finding clearly suggested that derivative 4a significantly inhibited the Aβ aggregation and had good peripheral anionic site-AChE binding affinity.
2.3.6. Molecular docking study
The molecular docking was performed using the Glide in extra precise (XP) mode on the crystal structures of AChE with co-crystallized ligannd donepezil (Friesner et al. 2006). The grid was described using the co-crystallized ligand donepezil. The AChE was redocked with the co-crystallized ligand. Both co-crystallized and docked donepezil binding sites were compared. It was discovered that the ‘RMSD’ difference between co-crystallized and docked donepezil was 0.657. Table 2 displays the results of molecular docking of all ligands 4a-4f against the AChE. The ligand 4a primarily interacts with Arg296 via hydrogen bonds, although it also interacts with Tyr337 and His447 via π-π stacking interactions. The potent molecule DPZ mainly interacts with Phe295, Trp86, Trp286, Tyr337 and one water molecules via hydrogen bonds, π-π interaction, and π-cation interactions.
Table 2
Depict the docking results and residual interactions of compound 4a-4f and DPZ.
|
S. NO. |
Compound Name |
docking score |
XP GScore |
glide gscore |
glide emodel |
Interactions |
||
|
Hydrogen bonding |
π-π interaction |
π-cation |
||||||
|
1 |
4a |
-6.462 |
-6.462 |
-6.462 |
-47.762 |
Arg296 |
Tyr337, His447 |
|
|
2 |
4b |
-6.821 |
-6.821 |
-6.821 |
-50.662 |
|
Trp86, His447, Tyr337 |
|
|
3 |
4c |
-4.99 |
-4.99 |
-4.99 |
-43.241 |
Tyr124, HOH |
Tyr124 |
|
|
4 |
4d |
-5.097 |
-5.097 |
-5.097 |
-43.259 |
Tyr124, HOH |
Tyr124 |
|
|
5 |
4e |
-8.61 |
-8.61 |
-8.61 |
-53.502 |
|
His447 |
|
|
6 |
4f |
-7.424 |
-7.424 |
-7.424 |
-63.968 |
|
His447 |
|
|
7 |
DPZ |
-16.598 |
-16.602 |
-16.602 |
-96.152 |
Phe295, HOH |
Trp286, Trp86 |
Tyr337, Trp86 |
2.3.7 Molecular dynamics simulations
Interaction of Donepezil and 4a with hAChE
During MD simualtions it was observed that the piperidine moiety of DPZ mainly interacts with three different residues Trp86, Tyr337, and Phe338 via π-cation inteactions (Figure6). The protein-ligand contact histogram and protein-ligand contact timeline (Figure S3) depict that the hydrophobic interactions of residues Trp86, Tyr337, and Phe338 with DPZ are stable and consistent. The benzyl moiety attached to piperidinealso makes a stable and consistent π-π stacking interaction with the Trp86. The dark color in protein-ligand contact timeline shows the multiple interaction of Trp86 with DPZ (Figure S3). Similarly, the dimethoxyindanone moiety of DPZ also intearcts with Trp286 and Phe295 via π-π stacking and hydrogen bond respectively (Figure 6).
The benzothiazole moiety present in the ligand 4a mainly interacts with Tyr337, Trp 86 and His 447 via hydrogen bonds and hydrophobic (π-π interaction) interactions(Figure 6). The protein-ligand contact histogram and timeline depict that intearction of residues Tyr337 (hydrogen bonds) and Trp86 (π-π interaction) with 4a are prominent, consistent and stable (Figure S4). The phenolic moiety of 4a also makes water mediated hydrogen bond and direct hydrogen bonds with Trp286 and Ser298, respectively (Figure 6).
Analysis of structural stability, compactness and binding free energy
The radius of gyration and root mean square deviation (RMSD) are important parameters to examine the structural stability of AChE and its complexes with various chemicals (Rg). The root mean square deviation plot of apo-AChE and its complex with the DPZ is shown in Figure 7 (4a). The RMSD changes for both complexes indicate that the simulation has reached a point of convergence. As previously noted in earlier studies, binding of ligands, namely DPZ, results in a considerable reduction in the RMSD value. [26,27] During the last few ns of simulations, the RMSD fluctuation of AChE decreased in the presence of 6a. There is significant decrease in average RMSD value of AChE in presence of both ligands. Figure 7 shows the significant alteration in the Rg of the AChE in presence and in absence of the compounds. The Rg is an main predictor of the protein compactness. The Rg plot (Figure 7) and average Rg analyzed from the whole trajectory indicate that there are no considerable and significant changes in Rg of AChE in presence of ligand as compared to apo-AChE.
The last 25 ns trajectory was used to compute the binding free energy between AChE and both ligands (DPZ and 4a). In table 13 summarises the final bindiing free energies & its numerous supporting terms for all simulated complexess. AChE-DPZ and AChE-4a complexes had average binding energies was found to be of -64.19 3.7 and -64.324.3 Kcal/mol, respectively.
Table 3
The average ΔGbind (Kcal/mol) and its contributing energy terms Donepezil and 4a against hAChE calculated from MD trajectories (last 25) ns.
|
Complex |
Avg. ΔGbind Coulomb1 |
Avg. ΔGbind Covalent2 |
Avg. ΔGbind Hbond3 |
Avg. ΔGbind Lipo4 |
Avg. ΔGbind Packing5 |
Avg. ΔGbindsolv GB6 |
Avg. ΔGbind vdW7 |
ΔGbindTotal8 |
|
AChE_DPZ |
-11.99 |
1.71 |
-0.47 |
-32.73 |
-5.57 |
32.11 |
-47.24 |
-64.19±3.7 |
|
AChE_4a |
-21.00 |
2.29 |
-1.33 |
-24.38 |
-5.79 |
29.40 |
-43.50 |
-64.32±4.3 |
1Coulomb energy, 2Covalent binding energy, 3Hydrogen bonding correction, 4Lipophilic energy, 5Pi-pi packing correction, 6Generalized Born electrostatic solvation energy, 7Van der Waals energy and 8Total binding free energy.
2.3.8. In-vitro blood-brain barrier permeation assay
For CNS active candidates, blood-brain barrier (BBB) permeation is the main selection criteria. All the developed analogs were tested for their BBB permeation assessment using parallel artificial membrane permeation assay (PAMPA-BBB) (Supporting figure S5). The permeation constant of two marketed drugs were used as a positive and negative control for BBB permeability (Pe). The Pevalues of compounds (4a-4f) are listed in Table 1. Among all the synthesized analogs, compound 4a, Pe = 8.36 ± 0.56 X 10-6 cm s-1) showed highest BBB permeability compared to all derivatives. Moreover, all the synthesized compounds passes BBB in-vitro, with good permeation potentials.
2.3.9. Drug likeness studies
All the developed derivatives along with their intermediate 3a-3c and 4a-4f were assessed for their drug-likeness property determination (Lipinski's rule of five) using admetSAR software (Table 4). Molecular weight, logP, no. of hydrogen donors and acceptors and rotatable bond were determined using admetSAR software (Table 4). Lipinski’s points can be summarized as follows.
1. Molecular weight (MW) should be 500 g/mol or less.
2. M logP:(n-octanol/water partition coefficient) should be 5 or lesser.
3. Topological polar surface area (TPSA) should be less than 140 A.
4. Number of H-bond acceptors (HBAs) should be 10 or below and number of H-bond donors
(HBDs) should be 5 or below.
5. Number of rotatable bonds (RBs) should be 10 or below
Form the data depicted in Table 4, we can clearly conclude that our developed compound presented good drug likeness profile.
Table 4
Theoretical prediction of physiochemical parameter of developed analogs.
|
Compound Code |
MW |
logPa |
TPSAb |
HBAc |
HBDd |
RBs |
|
3a |
284.31 |
2.98 |
55.77 |
4 |
1 |
5 |
|
3b |
284.31 |
3.19 |
55.77 |
4 |
1 |
5 |
|
3c |
254.28 |
3.39 |
46.53 |
3 |
1 |
4 |
|
4a |
445.54 |
5.41 |
67.17 |
6 |
1 |
5 |
|
4b |
445.54 |
5.38 |
67.19 |
6 |
1 |
5 |
|
4c |
445.54 |
5.80 |
67.19 |
6 |
1 |
5 |
|
4d |
445.54 |
5.78 |
67.19 |
6 |
1 |
5 |
|
4e |
415.52 |
5.79 |
57.96 |
5 |
1 |
4 |
|
4f |
415.52 |
5.76 |
57.96 |
5 |
1 |
4 |
2.3.9. Prediction of ADMET, blood-brain barrier (BBB) penetration properties.
Absorption, distribution, metabolism, and execration (ADME) is one of the major criteria in the development of orally active drug. The admetSAR server (http://lmmd.ecust.edu.cn:8000/predict/) was used to estimate the target compound's ADMET profile, BBB permeability, and acute toxicity using molecular modelling. Table 5 shows that the developed compound 4a has a fine ADMET profile & potential BBB permeability according to admetSAR.
Table 5
Theoretical prediction of ADMET profile, blood-brain barrier (BBB) penetration of 4a.
|
Compound |
Model |
Results |
Probability |
|
4a |
Plasma protein binding |
100% |
1.08 |
|
4a |
Blood-Brain Barrier |
+ |
0.9765 |
|
4a |
Caco-2 Permeability |
+ |
0.5287 |
|
4a |
Acute oral toxicity |
III |
0.6006 |
|
4a |
Fish Toxicity |
- |
0.9466 |
This study depicted the development of pyrazolo-benzothiazole derivatives as a multifunctional agent for the management of AD. All the developed derivatives were tested for their multifunctional property including ChE inhibition, antioxidant evaluation using DPPH assay, enzyme kinetics study and Aβ modulation study. Among all developed derivatives, 4acontaining 3,4-dimethoxy substitution was the most potent AChE inhibitor.SARstudy clearly revealed that di-methoxy substituted derivatives showed better activity compared to that of mono-substituted derivatives. DPPH assay clearly revealed that 4a significantly presented free radical scavenging activity. Aβ modulation experiment showed Aβ anti-aggregation property of 4a. Docking study showed that compound 4a correctly adjusted within the pocket of AChE via strong hydrogen bond and hydrophobic interactions. Finally, the current study clearly indicates that the pyrazolo-benzothiazole hybrids can be a potential core for further development of anti-AD agent (Supporting figure S6).
4.1 Chemistry
All the solvents used for the synthesis of the developed compounds were dried by distillation techniques before use. All the chemicals and reagents were obtained from the CDH chemicals (India), Qualigen (India), S.D. Fine Chemicals (India) and Finar chemicals (India). The progress of reactions was checked by thin-layer chromatography (TLC) on precoated silica gel 60 F254 (MerckKGaA) and were checked under UV light. Column chromatographic purifications were done using silica gel 60-120 mesh size (Avra synthesis, India). 1H nuclear magnetic resonance (1H-NMR) and 13C NMR spectra were measured on Bruker Advance, 500 MHz spectrometers with tetramethylsilane (TMS) as the internal standard. The NMR solvents used were CDCl3 or DMSO‑d6 as indicated. Chemical shifts were measured in ppm and coupling constants (J) were measured in Hz. The following abbreviations are used to describe peak splitting patterns when appropriate: d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublet, br = broad. Coupling constants J are reported in Hertz (Hz).
4.1.1. General procedure for synthesis of chalcone (3a-3c)
Into a stirring solution of 4-hydroxyacetophenone (2) (1.2 g, 8.81 mmol, 1.0 equiv.) in ethanol (20 mL), substituted benzaldehyde (1a-1c) (1.75 g, 10.57 mmol, 1.20 equiv.) were added. After 5 min, KOH (1.48 g, 26.43 mmol, 3.0 mmol) was added to the reaction mixture. The mixture was refluxed for 10-12 h. After cooling down to RT, the solution was acidified with dilute HCl and left for 2 hr. The obtained precipitate was filtered and washed with a mixture of water. The resulting yellow solid was crystalized using ethanol to yield compound 3a-3c as a solid yellow powder in good to high yield.
4.1.1.1. (E)-3-(3,4-dimethoxyphenyl)-1-(4-hydroxyphenyl)prop-2-en-1-one (3a)
Compound 3a was prepared according to general procedure mentioned above. 4-hydroxyacetophenone (2) (1.2 g, 8.81 mmol, 1.0 equiv.), 3, 4-methoxybenzaldehyde (1a) (1.75 g, 10.57 mmol, 1.2 equiv.), KOH (1.48 g, 26.43 mmol, 3.0 mmol) were mixed in 20 mL ethanol, and refluxed for 10 hr. Yellow solid powder (1.242 g, 81% yield), TLC (EtOAc:Hexane 30:70 v/v), Rf = 0.60. 1H NMR (500 MHz, DMSO-d6):δ10.46 (bs, 1H, -OH), 8.09 (d, J = 8.75 Hz, 2H, Ar-H), 7.81 (d, J = 15.50 Hz, 1H, -CH=CH-), 7.66 (d, J = 15.50 Hz, 1H,-CH=CH-), 7.52 (d, J = 5.50 Hz, 1H, Ar-H), 7.34 (dd, J1 = 8.5 Hz, J2 = 2.5 Hz, 1H,Ar-H), 6.99 (d, J = 6.50 Hz, 1H, Ar-H), 6.93 (d, J = 8.50 Hz, 2H, Ar-H), 3.86 (s, 3H, -OCH3), 3.80 (s, 3H, -OCH3).
4.1.1.2. (E)-3-(2,5-dimethoxyphenyl)-1-(4-hydroxyphenyl)prop-2-en-1-one (3b)
Compound 3b was prepared according to general procedure mentioned above. 4-hydroxyacetophenone (2) (1.2 g, 8.81 mmol, 1.0 equiv.), 2, 5-methoxybenzaldehyde (1b) (1.75g, 10.57 mmol, 1.2 equiv.), KOH (1.48 g, 26.43 mmol, 3.0 mmol) were mixed in 20 mL ethanol, and refluxed for 10 hr. Yellow solid powder (1.32 g, 78% yield), TLC (EtOAc:Hexane 35:65 v/v), Rf = 0.55. 1H NMR (500 MHz, DMSO-d6):δ10.32 (bs, 1H, -OH), 8.08 (d, J = 8.0 Hz, 2H, Ar-H), 7.99 (d, J = 16.0 Hz, 1H, -CH=CH-), 7.88 (d, J = 16.0 Hz, 1H, -CH=CH-), 7.53 (d, J = 6.0 Hz, 1H, Ar-H), 7.01-7.00 (m, 2H, Ar-H), 6.92 (d, J = 6.50 Hz, 2H, Ar-H), 3.83 (s, 3H, -OCH3), 3.79 (s, 3H, -OCH3).
4.1.1.3. (E)-1-(4-hydroxyphenyl)-3-(4-methoxyphenyl)prop-2-en-1-one (3c)
Compound 3c was prepared according to general procedure mentioned above. 4-hydroxyacetophenone (2) (1.2 g, 8.81 mmol, 1.0 equiv.), 4-methoxybenzaldehyde (1c) (1.42 g, 10.57 mmol, 1.2 equiv.), KOH (1.48 g, 26.43 mmol, 3.0 mmol) were mixed in 20 mL ethanol, and refluxed for 10 hr. Yellow solid (1.10 g, 76% yield), TLC (EtOAc:Hexane40:60 v/v), Rf = 0.50. 1H NMR (500 MHz, DMSO-d6):δ10.24 (bs, 1H, -OH), 8.01 (d, J = 7.5 Hz, 2H, Ar-H), 7.82 (d, J = 15.75 Hz, 1H, -CH=CH-), 7.78 (d, J = 15.75 Hz, 1H, -CH=CH-), 7.53 (d, J = 8.5 Hz, 2H, Ar-H), 7.11-7.07 (m, 2H, Ar-H), 6.98 (d, J = 8.5 Hz, 2H, Ar-H), 3.68 (s, 3H, -OCH3).
4.1.2. General procedure for synthesis of substituted 2-hydrazinobenzothaizole
To a 60 mL seal tube equipped with magnetic stirrer, 4-methylbenzo[d]thiazol-2-amine (1.64g, 10 mmol, 1.0 equiv.), ethylene glycol (30 ml), and hydrazine hydrate (1.6 mL, 50 mmol, 5 equiv.) were added. The reaction mixture was then heated at 150 oC for 10 hr. The progress of reaction was monitored by TLC followed by the addition of water (20 ml). The intermediates were then filter washed with water to get aforementioned product.
4.1.2.1. 2-hydrazinyl-6-methylbenzo[d]thiazole (6a)
Compound was prepared according to general procedure mentioned above. 6-methylbenzo[d]thiazol-2-amine (1.64g, 10 mmol, 1.0 equiv.), ethylene glycol (30 ml), and hydrazine hydrate (1.6 mL, 50mmol, 5 equiv.) were mixed and refluxed for 10 hr. White solid powder (1.72, 78% yield), TLC (EtOAc:Hexane 50:50 v/v), Rf = 0.55. 1H NMR (500 MHz, DMSO-d6):δ9.01 (bs, 1H, -NH), 7.49 (dd, J1 = 7.5 Hz, J2 = 2.5 Hz, 1H, Ar-H), 7.01 (d, J = 8.0 Hz, 1H, Ar-H), 6.88 (t, J = 6.5 Hz, 1H, Ar-H), 4.99 (s, 2H, -NH2), 2.51 (s, 3H, -CH3).
4.1.2.2. 2-hydrazinyl-4-methylbenzo[d]thiazole (6b)
Compound was prepared according to general procedure mentioned above. 4-methylbenzo[d]thiazol-2-amine (1.64 g, 10 mmol, 1.0 equiv.), ethylene glycol (30 ml), and hydrazine hydrate (1.6 mL, 50 mmol, 5 equiv.) were mixed and refluxed for 10 hr. White solid crystalline powder (1.8 g, 81% yield), TLC (EtOAc:Hexane 50:50 v/v), Rf = 0.60. 1H NMR (500 MHz, DMSO-d6): δ 8.86 (bs, 1H, -NH), 7.47 (s, 1H, Ar-H), 7.20 (d, J = 7.5 Hz, 1H, Ar-H), 7.01 (dd, J1 = 8.5 Hz, J2 = 2.0 Hz, 1H, Ar-H), 4.96 (s, 2H, -NH2), 2.50 (s, 3H, -CH3).
4.1.3. General procedure for synthesis of pyrazolo-benzothiazole derivatives
To a 30 mL seal tube equipped with magnetic stirrer, compound 3a-3c (1.0 equiv.), and compound 6a-6b (1.2 equiv.) were refluxed in n-butanol for 10-12h. The progress of reaction was monitored by TLC (EtOAc:Hexane, 1:1). The crude residues were then filter and subjected to column chromatography to get aforementioned product.
4.1.3.1.4-(5-(3,4-dimethoxyphenyl)-1-(6-methylbenzo[d]thiazol-2-yl)-4,5-dihydro-1H-pyrazol-3-yl)phenol (4a)

Compound was prepared according to general procedure mentioned above. (E)-3-(3,4-dimethoxyphenyl)-1-(4-hydroxyphenyl)prop-2-en-1-one(3a) (0.2 g, 0.703 mmol, 1.0 equiv.) and 2-hydrazinyl-6-methylbenzo[d]thiazole (6a) (0.126 g, 0.703 mmol, 1.0 equiv.) were refluxed in n-butanol for 10-12h. White solid crystalline powder (0.289 mg, 80% yield), TLC (EtOAc:Hexane 1:1 v/v), Rf = 0.60. 1H-NMR (500 MHz, DMSO) δ ppm: 7.60 (m, 3H, Ar-H), 7.06 (d, J = 7.3 Hz, 1H, Ar-H), 7.01 – 6.95 (m, 2H, Ar-H), 6.87 – 6.80 (m, 3H, Ar-H), 6.67 (d, J = 3.0 Hz, 1H, Ar-H), 5.84 (dd, J = 11.8, 5.3 Hz, 1H, -CH), 3.93 (q, 1H, -CH2), 3.78 (s, 3H, -OCH3), 3.62 (s, 4H, -OCH3, methylene -H), 2.31 (s, 3H, -CH3). 13C-NMR (126 MHz, DMSO) δ ppm: 161.98, 154.87, 153.51, 151.75, 151.17, 130.95,128.73, 128.62,126.82, 121.82, 119.06, 116.28, 114.72, 114.30,113.36, 113.03, 59.48, 56.82, 55.74, 18.11.
4.1.3.2.4-(5-(3,4-dimethoxyphenyl)-1-(4-methylbenzo[d]thiazol-2-yl)-4,5-dihydro-1H-pyrazol-3-yl)phenol (4b)

Compound was prepared according to general procedure mentioned above. (E)-3-(3,4-dimethoxyphenyl)-1-(4-hydroxyphenyl)prop-2-en-1-one(3a) (0.2 g, 0.703 mmol, 1.0 equiv.) and 2-hydrazinyl-4-methylbenzo[d]thiazole (6b) (0.126 g, 0.703 mmol, 1.0 equiv.) were refluxed in n-butanol for 10-12h. White solid crystalline powder (0.25 mg, 75% yield), TLC (EtOAc:Hexane 1:1 v/v), Rf = 0.60. 1H NMR (500 MHz, DMSO) δppm: 7.60 (dd, J = 17.7, 8.1 Hz, 3H, Ar-H), 7.06 (d, J = 7.3 Hz, 1H, Ar-H), 7.06 – 6.90 (m, 2H, Ar-H), 6.93 – 6.75 (m, 3H, Ar-H), 6.67 (d, J = 3.0 Hz, 1H), 5.84 (dd, J = 11.8, 5.3 Hz, 1H), 3.94 (d, J = 11.8 Hz, 2H, -CH2), 3.84 (d, J = 6.5 Hz, 3H, -OCH3), 3.62 (s, 3H, -OCH3), 2.31 (s, 3H, -CH3).13C NMR (126 MHz, CDCl3) δppm: 163.14, 161.19, 147.41, 146.74, 143.04, 130.69, 130.63, 127.80, 121.88, 115.28, 115.11, 114.80, 114.40, 109.92, 62.03, 55.98, 31.93, 29.70.
4.1.3.3. 4-(5-(2,5-dimethoxyphenyl)-1-(6-methylbenzo[d]thiazol-2-yl)-4,5-dihydro-1H-pyrazol-3-yl)phenol (4c)

Compound was prepared according to general procedure mentioned above. (E)-3-(2,5-dimethoxyphenyl)-1-(4-hydroxyphenyl)prop-2-en-1-one(3b) (0.2 g, 0.703 mmol, 1.0 equiv.) and 2-hydrazinyl-6-methylbenzo[d]thiazole (6a) (0.126 g, 0.703 mmol, 1.0 equiv.) were refluxed in n-butanol for 10-12h. White solid powder (0.26 mg, 76% yield), TLC (EtOAc:Hexane 1:1 v/v), Rf = 0.50. 1H-NMR (500 MHz, DMSO) δ ppm: 7.69 (d, J = 7.5 Hz, 2H, Ar-H), 7.52 (s, 1H, Ar-H), 7.12 (s, 1H, Ar-H), 7.01 (d, J = 7.5 Hz, 2H, Ar-H), 6.92 (s, 1H, Ar-H), 6.68– 6.62 (m, 2H, Ar-H), 6.57 (d, J = 3.0 Hz, 1H, Ar-H), 5.75 (dd, J = 8.5, 3.5 Hz, 1H, -CH), 4.01 (t, J = 6.5, 1H, -CH2), 3.63 (s, 3H, -OCH3), 3.51 (s, 4H, -OCH3, methylene -H), 2.39 (s, 3H, -CH3). 13C-NMR (126 MHz, DMSO) δ ppm: 160.63, 151.36, 149.68, 148.61, 147.38, 145.36,131.31, 125.18,122.63, 121.45, 118.74, 115.64, 112.31, 111.27,110.32, 110.17, 58.62, 57.21, 54.32, 19.64.
4.1.3.4. 4-(5-(2,5-dimethoxyphenyl)-1-(4-methylbenzo[d]thiazol-2-yl)-4,5-dihydro-1H-pyrazol-3-yl)phenol (4d)

Compound was prepared according to general procedure mentioned above. (E)-3-(2,5-dimethoxyphenyl)-1-(4-hydroxyphenyl)prop-2-en-1-one(3b) (0.2 g, 0.703 mmol, 1.0 equiv.) and 2-hydrazinyl-4-methylbenzo[d]thiazole (6b) (0.126 g, 0.703 mmol, 1.0 equiv.) were refluxed in n-butanol for 10-12h. White solid powder (0.275 mg, 80% yield), TLC (EtOAc:Hexane 1:1 v/v), Rf = 0.60. 1H-NMR (500 MHz, DMSO) δ ppm: 7.64 (d, J = 8.5 Hz, 2H, Ar-H), 7.50 (s, 1H, Ar-H), 7.08 (s, 1H, Ar-H), 6.99 (m, 3H, Ar-H), 6.71 (d, J = 8.5, 2H, Ar-H), 6.60 (d, J = 7.5 Hz, 1H, Ar-H), 5.76 (d, J = 8.5, 1H, -CH), 3.94 (d, J = 7.5, 2H, -CH2), 3.70 (s, 3H, -OCH3), 3.44 (s, 3H, -OCH3), 2.56 (s, 3H, -CH3). 13C-NMR (126 MHz, DMSO) δ ppm: 163.23, 155.36, 154.12, 149.62, 148.14, 147.65,141.74, 141.02,136.21, 127.32, 119.82, 116.84, 117.62, 114.61,109.51, 108.62, 60.32, 59.65, 55.41, 20.14.
4.1.3.5.4-(5-(4-methoxyphenyl)-1-(6-methylbenzo[d]thiazol-2-yl)-4,5-dihydro-1H-pyrazol-3-yl)phenol (4e)

Compound was prepared according to general procedure mentioned above. (E)-3-(4-methoxyphenyl)-1-(4-hydroxyphenyl)prop-2-en-1-one(3c) (0.2 g, 0.703 mmol, 1.0 equiv.) and 2-hydrazinyl-6-methylbenzo[d]thiazole (6a) (0.126 g, 0.703 mmol, 1.0 equiv.) were refluxed in n-butanol for 10-12h. White solid crystalline powder (0.245 mg, 75% yield), TLC (EtOAc:Hexane 1:1 v/v), Rf = 0.60. 1H-NMR (500 MHz, DMSO) δ ppm: 7.54 (d, J = 8.5 Hz, 2H, Ar-H), 7.44 (d, J = 7.5 Hz, 2H, Ar-H), 7.05 (d, J = 7.5, 2H, Ar-H), 6.94 (s, 1H, Ar-H), 6.66 (d, J = 7.5, 2H, Ar-H), 6.47 (d, J = 7.5 Hz, 1H, Ar-H), 6.41 (s, 1H, Ar-H), 5.80 (d, J = 6.5, 1H, -CH), 4.12 (d, J = 6.5, 2H, -CH2), 3.61 (s, 3H, -OCH3), 3.51 (s, 3H, -OCH3), 2.41 (s, 3H, -CH3). 13C-NMR (126 MHz, DMSO) δ ppm: 161.32, 158.14, 151.32, 147.52, 144.65, 143.84,140.66, 137.63,135.41, 125.62, 118.14, 115.11, 114.21, 113.21, 106.44, 57.32, 56.44, 53.94, 18.65.
4.1.3.6.4-(5-(4-methoxyphenyl)-1-(4-methylbenzo[d]thiazol-2-yl)-4,5-dihydro-1H-pyrazol-3-yl)phenol (4f)

Compound was prepared according to general procedure mentioned above. (E)-3-(4-methoxyphenyl)-1-(4-hydroxyphenyl)prop-2-en-1-one(3c) (0.2 g, 0.703 mmol, 1.0 equiv.) and 2-hydrazinyl-4-methylbenzo[d]thiazole (6b) (0.126 g, 0.703 mmol, 1.0 equiv.) were refluxed in n-butanol for 10-12h. White solid powder (0.29 mg, 82% yield), TLC (EtOAc:Hexane 1:1 v/v), Rf = 0.55. 1H-NMR (500 MHz, DMSO) δ ppm: 7.61 (d, J = 7.5 Hz, 2H, Ar-H), 7.56 (d, J = 6.5 Hz, 2H, Ar-H), 6.99 (d, J = 8.5, 2H, Ar-H), 6.91 (m, 3H, Ar-H), 6.40 (d, J = 8.5 Hz, 1H, Ar-H), 6.38 (s, 1H, Ar-H), 5.84 (d, J = 7.5, 1H, -CH), 4.21 (d, J = 7.0, 2H, -CH2), 3.66 (s, 3H, -OCH3), 3.55 (s, 3H, -OCH3), 2.44 (s, 3H, -CH3). 13C-NMR (126 MHz, DMSO) δ ppm: 160.32, 159.74, 154.12, 150.21, 147.63, 147.15,138.15, 133.12,131.50, 124.32, 116.50, 111.16, 110.74, 109.84, 108.63, 60.84, 58.62, 55.12, 20.32.
4.2. Biological evaluation
4.2.1. Determination of IC50 values
Cholinesterase inhibitory activity of developed molecules was assessed calorimetrically using modified Ellmann method.[28] ChE selectively catalyzes the breakage of acetylthiocholine to thiocholine and acetic acid, which reduces the 5,5-dithio-bis-(2- nitrobenzoic acid) (DTNB) to produces yellow color intermediate that can be spotted colorimetrically at 413 nm. Human acetylcholinesterase (hAChE) (CAS No. 9000-81-1), butyrylcholinesterase (eqBChE) (CAS NO. 9001-08-5), 5,5-dithiobis-2-nitrobenzoic acid (DTNB, CAS No. 69-78-3), acetylthiocholine iodide (ATCI, CAS No. 1866-15-5) and butyrylthiocholine iodide (BTCI, CAS No. 1866-16-6) werebrought from Sigma Aldrich, and Spectrochem chemicals respectively. All the experiments were performed in 50 mM phosphate buffer at pH 7.4.
Briefly, 50 mL of AChE (0.22 U/mL, initial concentration) and 10 μL of the test or standard compound were incubated in 96-well plates at room temperature for 30 min. Furthermore, 30 μL of the substrate viz. ATCI (1.5 mM) was added. After 30 minutes, 160 μL of DTNB (0.15 mM) was added to it and then absorbance was measured at 413 nm wavelength using microplate reader. Each assay was performed in triplicate. The blank contained all components except enzyme. The inhibition percent was calculated by the following expression: [(AcAi)/Ac] X100, where Ai and Ac are the absorbance obtained for BChE in the presence and absence of inhibitors.
The in-vitro BChE inhibition experiment was performed using the same procedure as described above. Briefly, 50 μL of BChE (0.06 U/mL) and 10 μL of the test or reference compound were incubated in 96-well plates at RT for 30 min. Thereafter, 30 μL of the substrate viz. BTCI (15 mM) was added and the solution. After 30 minutes, 160 μL of DTNB (1.5 mM) was added to it and the absorbance was measured at 413 nm wavelength using a 96-well microplate reader. The inhibition percent was calculated by the following expression: [(AcAi)/Ac] X100, where Ai and Ac are the absorbance obtained for BChE in the presence and absence of inhibitors.
4.2.2. Kinetic characterization of AChE and BChE inhibition
In order to evaluate the mechanism of action of 4a, reciprocal plots of 1/[V] versus 1/[S] were constructed using six different concentrations of the substrate ATCI (from 0.5, 1.0, 1.5, 2.0, 2.5, and 3.0 mM for hAChE) by using the modified Ellman method.[28] Briefly, compound 4a (10μL) at different concentrations (5μM, 10μM and 20 μM) was pre-incubated with hAChE (50 μLof 0.22 U/mL at RT for 30 minutes, followed by the addition of 30 μL of the substrate at different concentrations. The kinetic characterization of the hydrolysis of ATCI catalyzed by AChE is done spectrometrically using a 96-well microplate reader at 413 nm.
4.2.3. DPPH radical-scavenging potency
The DPPH (2,2-diphenyl-1-picryl-hydrazyl-hydrate) free radical scavenging method is an simple and most convenient antioxidant assay based on reduction of DPPH radical. DPPH was purchased for Sigma-Aldrich (Merck, CAS No. 1898-66-4). All the experiments were carried out in biology grade methanol (for poor solubility ethanol can be used). Eight different concentrations 1, 10, 20, 40, 80, 100, 160, and 200 μM of test sample were used. In brief, 75 μL of different concentrations of the test sample were added to a 96-well plate. Then, 75 μL of DPPH (200 μM) solution was added to it. Finally, a 96-well microplate was allowed to stand at RT for 30 minutes, followed by absorbance at 520 nm using microplate reader. The radical scavenging capacity was determined using the equation % radical scavenging activity = [(absorbance of control-absorbance of the test)/absorbance of control] X 100. All the experiments were performed in triplicate.
4.2.4. PAMPA-BBB Assay
Brain permeability prediction of the developed compound was assessed by in-vitro PAMPA-BBB assay reported method from Di et al..[29] The protocol involved the coating of porcine brain lipid solution dissolved in dodecane (5 μL) on filter membrane of donar microplate. The 500 μM concentration of test compound was prepared in phosphate buffer (pH = 7.4). The donor and acceptor microplates were filled with 500 μM (200 μL) of compound and 300 μL of phosphate buffer respectively. The assembly of acceptor and donor was sandwiched with each other and incubated at RT for 16 h). After incubation, diffusion of the test compounds in the acceptor plate was calculated by determining the absorbance spectrophotometrically. The experiment was conducted in triplicate.
4.2.4. Inhibition of Aβ Aggregation.
Aβ peptide (CAS No. 107761-42-2), molecular biology grade DMSO (CAS No. 67-68-5), Phosphate buffer saline (PBS) was procured from Sigma-Aldrich and HiMedia respectively. Aβ peptide 0.1 mg was dissolved in 100 μL of molecular biology grade DMSO, and aliquot in five different vials. The test compounds were prepared in molecular biology grade DMSO and PBS pH 7.4 (DMSO ≤ 1% w/v final concentration). Two different ratios of Aβ peptide and 4a were evaluated (10:10 μM, and 10:20 μM respectively).
For self-induced anti-Aβ aggregation assay, the mixture of Aβ peptide (20 μM) Aβ peptide (40 μM) in PBS pH 7.4 in the presence or absence of inhibitor (20 μM, and 40 μM) was incubated (37 °C, 48 h) followed by addition of 100 μM of thioflavin T (10μM). The fluorescence intensity was measured at excitation (λex = 450 nm) and emission (λemission = 485 nm) wavelengths. The anti-Aβ aggregatory potential was calculated as percentage inhibition following an expression: [100 − (Fi/Fo × 100)]; and NFI = Fi/Fo. The Fi and Fo are the fluorescence intensities in the presence or absence of inhibitor, respectively.
4.2.5. Molecular docking
The 3D crystal structure of hAChE in complex with DPZ (PDB ID- 4EY7) was retrieved from the Brookhaven protein data bank.[30, 31] Protein Preparation Wizard of Schrodinger software package (Schrodinger, LLC, New York, NY) was used to prepare proteins. This step includes removal of water beyond 5 Å from the HET group, addition of missing hydrogen, optimization of orientations of hydroxyl and amino groups, assignment of right bond orders, and the determination of ionization of amino acids using ProtAssign utility. The resulting structures were further subjected to restrained minimization with cutoff root mean square deviation (RMSD) of 0.3 Å. Finally, the prepared complexes were further used for molecular docking and MD simulation study. All the small molecules were drawn using 2D sketcher and were subjected to ligand preparation using the LigPrep module of Schrodinger software package (Schrodinger, LLC, New York, NY). The different possible ionization states for ligands were generated at the physiological pH (7.0 ± 2), and OPLS4 force field was used to minimize the ligands. Finally, docking of all ligands was performed by the Glide module of the Schrodinger software package (Schrodinger, LLC, New York, NY) using standard operating procedures with the extra precision (XP) protocol.[32]
4.2.6 Molecular Dynamics Simulation
All-atom MD simulations were performed using the desmond-v6.6 module of Schrödinger Software Package (Schrödinger, LLC, New York, NY).[33] The system builder panel was used to prepare the initial systems for MD simulations. The apo-AChE and both docked complexes (AChE-DPZ, an AChE-4a) were placed in a cubic box of 1.0 nm size. The boxes were solvated with TIP3P water models and charged systems were neutralized using counter ions (Na+ or Cl- ions).[34] An ionic strength of 0.15 M was maintained by adding Na+ and Cl- ions to all the systems. Further, the solvated systems were minimized and equilibrated under NPT ensemble using the default protocol of Desmond. It includes a total of nine stages, among which there are two minimization and four short simulations (equilibration phase) steps.[35] All minimized and equilibrated systems were subjected to MD run with periodic boundary conditions in NPT ensemble using OPLS4 force field parameter for 100 ns.[36] During the simulation, the pressure (1 atm) and temperature (300 K) of the systems were maintained by Martyna–Tobias–Klein barostat and Nose–Hoover Chain thermostat, respectively.[37-40] The binding energy between the AChE and ligands (DPZ & 4a) was calculated using the inbuilt script thermal_mmgbsa.py.[41, 42] The binding energy was calculated from the last 25 ns of trajectory at an interval of 50 ps for both systems ((AChE-DPZ, an AChE-4a).
Acknowledgements
Harish Kumar would like to thank the Chitkara University for supporting this work.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supplementary materials
The data that support the findings of this study are available in the Supporting Information Material of this paper.
- M.A. DeTure, D.W. Dickson, The neuropathological diagnosis of Alzheimer’s disease, Molecular Neurodegeneration, 14 (2019) 32.
- P. Zhang, S. Xu, Z. Zhu, J. Xu, Multi-target design strategies for the improved treatment of Alzheimer's disease, European Journal of Medicinal Chemistry, 176 (2019) 228-247.
- Y.P. Singh, N. Kumar, K. Priya, B.S. Chauhan, G. Shankar, S. Kumar, G.K. Singh, S. Srikrishna, P. Garg, G. Singh, G. Rai, G. Modi, Exploration of Neuroprotective Properties of a Naturally Inspired Multifunctional Molecule (F24) against Oxidative Stress and Amyloid β Induced Neurotoxicity in Alzheimer’s Disease Models, ACS Chemical Neuroscience, 13 (2022) 27-42.
- M. Sadafi Kohnehshahri, G. Chehardoli, M. Bahiraei, T. Akbarzadeh, A. Ranjbar, A. Rastegari, Z. Najafi, Novel tacrine-based acetylcholinesterase inhibitors as potential agents for the treatment of Alzheimer’s disease: Quinolotacrine hybrids, Molecular Diversity, 26 (2022) 489-503.
- A. Rastegari, M. Safavi, F. Vafadarnejad, Z. Najafi, R. Hariri, S.N.A. Bukhari, A. Iraji, N. Edraki, O. Firuzi, M. Saeedi, M. Mahdavi, T. Akbarzadeh, Synthesis and evaluation of novel arylisoxazoles linked to tacrine moiety: in vitro and in vivo biological activities against Alzheimer’s disease, Molecular Diversity, 26 (2022) 409-428.
- M. Singh, M. Kaur, N. Chadha, O. Silakari, Hybrids: a new paradigm to treat Alzheimer’s disease, Molecular Diversity, 20 (2016) 271-297.
- M. Yazdani, N. Edraki, R. Badri, M. Khoshneviszadeh, A. Iraji, O. Firuzi, 5,6-Diphenyl triazine-thio methyl triazole hybrid as a new Alzheimer’s disease modifying agents, Molecular Diversity, 24 (2020) 641-654.
- Y.P. Singh, A. Pandey, S. Vishwakarma, G. Modi, A review on iron chelators as potential therapeutic agents for the treatment of Alzheimer’s and Parkinson’s diseases, Molecular Diversity, 23 (2019) 509-526.
- M. Mehrazar, M. Hassankalhori, M. Toolabi, F. Goli, S. Moghimi, H. Nadri, S.N.A. Bukhari, L. Firoozpour, A. Foroumadi, Design and synthesis of benzodiazepine-1,2,3-triazole hybrid derivatives as selective butyrylcholinesterase inhibitors, Molecular Diversity, 24 (2020) 997-1013.
- M. Singh, M. Kaur, H. Kukreja, R. Chugh, O. Silakari, D. Singh, Acetylcholinesterase inhibitors as Alzheimer therapy: from nerve toxins to neuroprotection, Eur J Med Chem, 70 (2013) 165-188.
- J. Hroudová, N. Singh, Z. Fišar, K.K. Ghosh, Progress in drug development for Alzheimer's disease: An overview in relation to mitochondrial energy metabolism, European Journal of Medicinal Chemistry, 121 (2016) 774-784.
- M. Shidore, J. Machhi, K. Shingala, P. Murumkar, M.K. Sharma, N. Agrawal, A. Tripathi, Z. Parikh, P. Pillai, M.R. Yadav, Benzylpiperidine-Linked Diarylthiazoles as Potential Anti-Alzheimer’s Agents: Synthesis and Biological Evaluation, Journal of Medicinal Chemistry, 59 (2016) 5823-5846.
- D.V. Patel, N.R. Patel, A.M. Kanhed, S.P. Patel, A. Sinha, D.D. Kansara, A.R. Mecwan, S.B. Patel, P.N. Upadhyay, K.B. Patel, D.B. Shah, N.K. Prajapati, P.R. Murumkar, K.V. Patel, M.R. Yadav, Novel Multitarget Directed Triazinoindole Derivatives as Anti-Alzheimer Agents, ACS Chemical Neuroscience, 10 (2019) 3635-3661.
- R. Farina, L. Pisani, M. Catto, O. Nicolotti, D. Gadaleta, N. Denora, R. Soto-Otero, E. Mendez-Alvarez, C.S. Passos, G. Muncipinto, C.D. Altomare, A. Nurisso, P.-A. Carrupt, A. Carotti, Structure-Based Design and Optimization of Multitarget-Directed 2H-Chromen-2-one Derivatives as Potent Inhibitors of Monoamine Oxidase B and Cholinesterases, Journal of Medicinal Chemistry, 58 (2015) 5561-5578.
- C.N. Pope, S. Brimijoin, Cholinesterases and the fine line between poison and remedy, Biochem Pharmacol, 153 (2018) 205-216.
- E. Tönnies, E. Trushina, Oxidative Stress, Synaptic Dysfunction, and Alzheimer's Disease, J Alzheimers Dis, 57 (2017) 1105-1121.
- D.B. Zorov, M. Juhaszova, S.J. Sollott, Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release, Physiol Rev, 94 (2014) 909-950.
- Z. Rozmer, P. Perjesi, Naturally occurring chalcones and their biological activities, Phytochemistry Reviews, 15 (2014).
- A. Rammohan, S. Julakanti, S. Gundala, Chittluri, N. Rao, Grigory, G. Zyryanov, Chalcone synthesis, properties and medicinal applications: a review, Environmental Chemistry Letters, 18 (2020).
- B. Salehi, C. Quispe, I. Chamkhi, N. El Omari, A. Balahbib, J. Sharifi-Rad, A. Bouyahya, M. Akram, M. Iqbal, A.O. Docea, C. Caruntu, G. Leyva-Gómez, A. Dey, M. Martorell, D. Calina, V. López, F. Les, Pharmacological Properties of Chalcones: A Review of Preclinical Including Molecular Mechanisms and Clinical Evidence, Front Pharmacol, 11 (2021) 592654-592654.
- X. Li, Y. Yu, Z. Tu, Pyrazole Scaffold Synthesis, Functionalization, and Applications in Alzheimer's Disease and Parkinson's Disease Treatment (2011-2020), Molecules, 26 (2021) 1202.
- F. Türkan, A. Cetin, M. Karaman, I. Gülçin, Synthesis, Biological Evaluation and Molecular Docking of Novel Pyrazole Derivatives as Potent Carbonic Anhydrase and Acetylcholinesterase Inhibitors, Bioorganic Chemistry, 86 (2019).
- K.M. Kasiotis, E.N. Tzanetou, S.A. Haroutounian, Pyrazoles as potential anti-angiogenesis agents: a contemporary overview, Frontiers in Chemistry, 2 (2014).
- K. Karrouchi, S. Radi, Y. Ramli, J. Taoufik, Y.N. Mabkhot, F.A. Al-Aizari, M.h. Ansar, Synthesis and Pharmacological Activities of Pyrazole Derivatives: A Review, Molecules, 23 (2018) 134.
- B. Uttara, A.V. Singh, P. Zamboni, R.T. Mahajan, Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options, Curr Neuropharmacol, 7 (2009) 65-74.
- N. Kumar, A. Gahlawat, R.N. Kumar, Y.P. Singh, G. Modi, P. Garg, Drug repurposing for Alzheimer’s disease: in silico and in vitro investigation of FDA-approved drugs as acetylcholinesterase inhibitors, Journal of Biomolecular Structure and Dynamics, (2020) 1-15.
- Y.P. Singh, G. Shankar, S. Jahan, G. Singh, N. Kumar, A. Barik, P. Upadhyay, L. Singh, K. Kamble, G.K. Singh, Further SAR studies on natural template based neuroprotective molecules for the treatment of Alzheimer’s disease, Bioorganic & Medicinal Chemistry, 46 (2021) 116385.
- P.W. Riddles, R.L. Blakeley, B. Zerner, Ellman's reagent: 5,5′-dithiobis(2-nitrobenzoic acid)—a reexamination, Analytical Biochemistry, 94 (1979) 75-81.
- L. Di, E.H. Kerns, K. Fan, O.J. McConnell, G.T. Carter, High throughput artificial membrane permeability assay for blood-brain barrier, Eur J Med Chem, 38 (2003) 223-232.
- H. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. Bhat, H. Weissig, I. Shindyalov, P. Bourne, P. Rose, A. Prlic, RCSB Protein Data Bank: Structural biology views for basic and applied research, Nucleic Acids Res, 28 (2000) 235-242.
- J. Cheung, M.J. Rudolph, F. Burshteyn, M.S. Cassidy, E.N. Gary, J. Love, M.C. Franklin, J.J. Height, Structures of human acetylcholinesterase in complex with pharmacologically important ligands, Journal of medicinal chemistry, 55 (2012) 10282-10286.
- R.A. Friesner, R.B. Murphy, M.P. Repasky, L.L. Frye, J.R. Greenwood, T.A. Halgren, P.C. Sanschagrin, D.T. Mainz, Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein− ligand complexes, Journal of medicinal chemistry, 49 (2006) 6177-6196.
- K.J. Bowers, D.E. Chow, H. Xu, R.O. Dror, M.P. Eastwood, B.A. Gregersen, J.L. Klepeis, I. Kolossvary, M.A. Moraes, F.D. Sacerdoti, Scalable algorithms for molecular dynamics simulations on commodity clusters, in: SC'06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, IEEE, 2006, pp. 43-43.
- P. Mark, L. Nilsson, Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K, The Journal of Physical Chemistry A, 105 (2001) 9954-9960.
- F.A. Samad, B.A. Suliman, S.H. Basha, T. Manivasagam, M.M. Essa, A comprehensive In Silico analysis on the structural and functional impact of SNPs in the congenital heart defects associated with NKX2-5 gene—A molecular dynamic simulation approach, PloS one, 11 (2016).
- D. Shivakumar, J. Williams, Y. Wu, W. Damm, J. Shelley, W. Sherman, Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field, Journal of chemical theory and computation, 6 (2010) 1509-1519.
- S. Nosé, A unified formulation of the constant temperature molecular dynamics methods, The Journal of chemical physics, 81 (1984) 511-519.
- D.J. Evans, B.L. Holian, The nose–hoover thermostat, The Journal of chemical physics, 83 (1985) 4069-4074.
- W.G. Hoover, Canonical dynamics: Equilibrium phase-space distributions, Physical review A, 31 (1985) 1695.
- K. Cho, J. Joannopoulos, L. Kleinman, Constant-temperature molecular dynamics with momentum conservation, Physical Review E, 47 (1993) 3145.
- P.D. Lyne, M.L. Lamb, J.C. Saeh, Accurate prediction of the relative potencies of members of a series of kinase inhibitors using molecular docking and MM-GBSA scoring, Journal of medicinal chemistry, 49 (2006) 4805-4808.
- S. Genheden, U. Ryde, The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities, Expert opinion on drug discovery, 10 (2015) 449-461.
Scheme 1 is available in the Supplementary Files section.
- Scheme1.png
Scheme 1. Synthesis of pyrazolo-benzothiazole analogues 4a-4f.
- SupportingInformation1.docx
{kind=link}