Plant material and phenotypic data
A diverse panel of 203 homozygous Chinese semi-winter inbred lines broadly encompassed the allelic variability in the Asian B. napus gene pool. The materials (Additional file 5: Table S1) were obtained as self-pollinated seeds from Southwest University, Chongqing, China, where they represent part of a breeding program spanning genetic diversity from the broader Asian gene pool. This population has been used previously in genomic diversity analysis [22] and GWAS of the chlorophyll content [25]. Field trials were conducted by applying an unreplicated complete randomized design in Chongqing in 2013, 2014 and 2015 (designated CQ-2013, CQ-2014, and CQ-2015, respectively). Each line was grown in a three-row plot with 10 homozygous inbred plants in each row. Self-pollinated seeds were collected from each line. About 5 g of dry mature seeds of each line (selected three strains, and each strain measured two times) were analyzed for oleic acid (C18:1) content by near-infrared spectroscopy (NIRS; Foss NIR Systems Inc., USA), each line selected three strains, and each strain measured two times.
The phenotypic analysis results, including the phenotypic distribution, mean value, standard deviation, correlation coefficient, and minimum and maximum values of oleic acid from the 203 accessions, were calculated and analyzed with the R packages Hmisc [41] and Psych [42]. Variance components were estimated via restricted maximum likelihood- using the statistical software package SPSS Statistics for Windows Version 22.0 (IBM Corp., Armonk, NY, USA) , and broad-sense heritability (H2) for the oleic acid content was calculated, across the environments and in each environment, using the following equation [43] : (see Equation 1 in the Supplementary Files)
where the genotypic and residual variance components are represented by and , respectively, and estimates of the residual variance were divided by the number (environments) .
Genotypic data
The 203 accessions were genotyped using the Brassica 60 K Illumina® Infinium SNP array (IlluminaInc., San Diego, CA, USA) by TraitGenetics GmbH (Gatersleben, Germany). We applied a filtering for single-copy BLAST hits that were physically assignable to the B. napus “Darmor-bzh” reference genome assembly version 4.1 [44] (http://www.genoscope.cns.fr/brassicanapus/data/). Only markers that exhibited at least 95% sequence identity and no gaps in their 50-bp probe sequence were retained, resulting in 28,698 unique, locus-specific SNPs. In an additional preprocessing step, all markers with more than 10% missing values and a minor allele frequency below 5% were excluded. A total of 24,338 high-quality, single-locus, SNP markers with a minor allele frequency (MAF) ≥0.05 were used for the GWAS (Additional file 8: Table S5).
Genome-wide association analysis
A detailed description of the genetic composition and population structure of the diversity set was provided by Qian et al. [22]. Relative kinship analysis was performed using the TASSEL 5.0 software [45]. Marker–trait associations were examined by applying the TASSEL 5.0 software with the Q + K mixed linear model as proposed by Yu et al. [46]. The mixed linear model was as follows: (see Equation 2 in the Supplementary Files)
The model was used to test associations between the SNPs and phenotypes, where y is the vector of phenotypic observations, μ is the overall mean, α is a vector of the fixed allelic effects associated with the SNP under investigation, v is a vector of fixed population effects, u is a vector of random genetic background effects, and e is the vector of residuals. The matrix P contains information on population structure, using the first five principal components as fixed factors to adjust the model for population stratification, while S and Z are incidence matrices relating y to a and u, respectively. The variances of the random effects u and e are assumed to be normally distributed with u ~ N(0, ) and e ~ N (0, ), where G is a 203×203 genomic relationship matrix and R is a 203×203 matrix in which the off-diagonal elements are 0 and the diagonal elements are the reciprocal of the number of phenotypic observations per individual. The G matrix was computed according to the first method proposed by VanRaden et al. [47]. Since every genotype was tested only once per environment and GWASs were performed for all the environment–trait combinations separately, R corresponds to the identity matrix I.
The observed P values from the marker-trait associations were used to display Q–Q and Manhattan plots using the R package qqman [48]. The critical P-value for assessing the significance of SNP-trait associations was calculated separately for the oleic acid content based on the false discovery rate (FDR) [498]. An FDR<0.05 was used to identify significant associations with the oleic acid content at a cut-off value of -log10(P) = 4.
50 accession resequencing analysis
Genomic DNA was extracted from fresh leaves of each plant using the cetyltrimethyl ammonium bromide (CTAB) method and sequenced using the Illumina HiseqTM 4000 (Illumina Co., Inc., San Diego, CA, USA) with a 5X sequencing depth and 125-bp paired-end sequencing length. Library preparation and sequencing were carried out at the Biomarker Technologies Corporation (Beijing, China). SNPs detected among the accessions that have described by Dong et al. [50]. SNPs with a missing call rate >0.6 were excluded. The remaining SNPs with missing call rates below 0.6 were filled using the software “beagle” v4.1 [51] (https://faculty.washington.edu/browning/beagle/beagle.html). SNP loci with a heterozygous rate greater than 25% and a MAF less than 0.05 were removed. Ultimately, we obtained 532,005 high-quality SNPs that were used to analysis LD among candidate genes on significant association haplotype region.
Phenotypic correlations with haplotype diversity groups
Significant haplotype blocks were identified using the R package LD heatmap [52], with haplotypes defined across regions of homozygous markers with LD (r2) >0.50 between the first and last markers in the block. Haplotype alleles with a frequency >0.01 were used for the comparative phenotype analysis. A two-sample t-test (assuming unequal variance) was used to test for significant phenotypic differences in the oleic acid content among the haplotype alleles.
Gene content in the haplotype block regions
Annotated genes within the oleic acid content-associated haplotype regions were extracted from the B. napus Darmor-bzh reference genome v. 4.1 [44]. To verify the most likely gene functions, we determined annotations of the closest orthologous Arabidopsis thaliana genes by BLAST matching against the Arabidopsis genome database (http://www.arabidopsis.org/).
Transcriptome sequencing
Thirteen of 50 resequencing accessions were selected for the sampling of siliques for transcriptome analysis fifteen days after pollination. These 13 accessions were selected based on different oil contents (i.e. 4 accessions with lower oil content, 4 with medium oil content and 5 with higher oil content). The siliques were immediately frozen in liquid nitrogen and stored at -80°C prior to RNA extraction. At least 3 µg of total RNA was used for each accession to construct paired-end sequencing libraries according to the manufacturer’s instructions (Illumina Inc.). Paired-end reads (125 bp) were evaluated using the Illumina HiSeq 2500 platform. The raw reads were filtered to remove sequencing adapters and low-quality reads (>10% unknown bases or >50% of bases with a quality <10) and generate “clean” reads that were subsequently aligned to the Darmor-bzh reference genome v.4.1 [44] using HISAT 2 (http://ccb.jhu.edu/software/hisat/index.shtml). The gene expression levels were calculated using HTSeq 0.6.1 [53] (https://pypi.python.org/pypi/HTSeq). To screen differentially expressed genes (DEGs), we used a q-value <0.05 and an absolute value of log2 (fold change) normalized to >1 based on the R package DEGSeq v1.22.0 [54].
Co-expression analysis
The co-expression edges were calculated, and a soft threshold value of 0.9 was chosen in the WGCNA R package [55]. Genes with Pearson Correlation Coefficients (PCCs) greater than or equal to 0.55 were used for co-expression network construction by CYTOSCAPE3.6 [56].
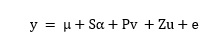{kind=link}
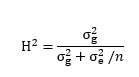{kind=link}