The high-risk screening for FD in selected patient cohorts has been reported. Doheny et al. [23] reanalyzed studies related to hemodialysis (27 reports, 23,954 males, and 12,866 females), left ventricular hypertrophy (LVH) and/or hypertrophic cardiomyopathy (17 reports, 4,054 males, and 1,437 females), and ischemic or cryptogenic strokes (16 studies, 3,904 males, and 2,074 females). The revised prevalence was estimated as 0.21% for male and 0.15% for female hemodialysis patients, 0.94% for male and 0.90% for female cardiac patients, and 0.13% for male and 0.14% for female stroke patients. In the current study, the prevalence was estimated as 0.42% (male: 0.45%, female: 0.38%) in the renal manifestation group (a), 0.94% (male: 0.77%, female: 1.33%) in the cardiac manifestation group (b), and 0.22% (male: 0.20%, female: 0.27%) in the central neurological manifestation group (c), which are comparable to those of previous reports.
The prevalence of FD in the peripheral neurological manifestation group (d) was second highest at 4.37% (male: 4.98%, female: 3.50%), whereas the patients’ age (male: median 13 [IQR: 11-18.5] years old, female: median 13 [IQR: 9-25] years old) was younger than the other groups. Therefore, manifestations such as limb pain, acroparesthesia, clustered angiokeratoma, cornea verticillata, and hypo- or anhidrosis, could be useful in the detection of Fabry patients. Politei et al. [24] has recommended that the cause of pain should be diagnosed early in unrecognized or newly diagnosed Fabry patients in order to improve treatment possibilities. FD experts consider that, regardless of gender or age, pain related to FD could be an early indication to commence ERT before potentially irreversible organ damage, to the kidneys, heart, or brain, prevails. However, a study conducted in Russia by Namazova-Baranova et al. [25] reported that no Fabry patients were detected from 214 patients (110 males and 104 females) with chronic limb pain. Moreover, the genetic, epidemiological, and ethnical information related to Russian Fabry patients are insufficient and future studies and information related to FD in Russia are required.
The prevalence of FD in individuals with a family history (e) was the highest at 23.40% (male: 12.49%, female: 33.02%). The GLA sequencing for individuals who had a family history of FD demonstrated its usefulness in detecting undiagnosed or pre-symptomatic Fabry patients. Therefore, when patients experience FD-related symptoms, clinicians should confirm the presence of a family history of FD and, if applicable, whether similar symptoms developed.
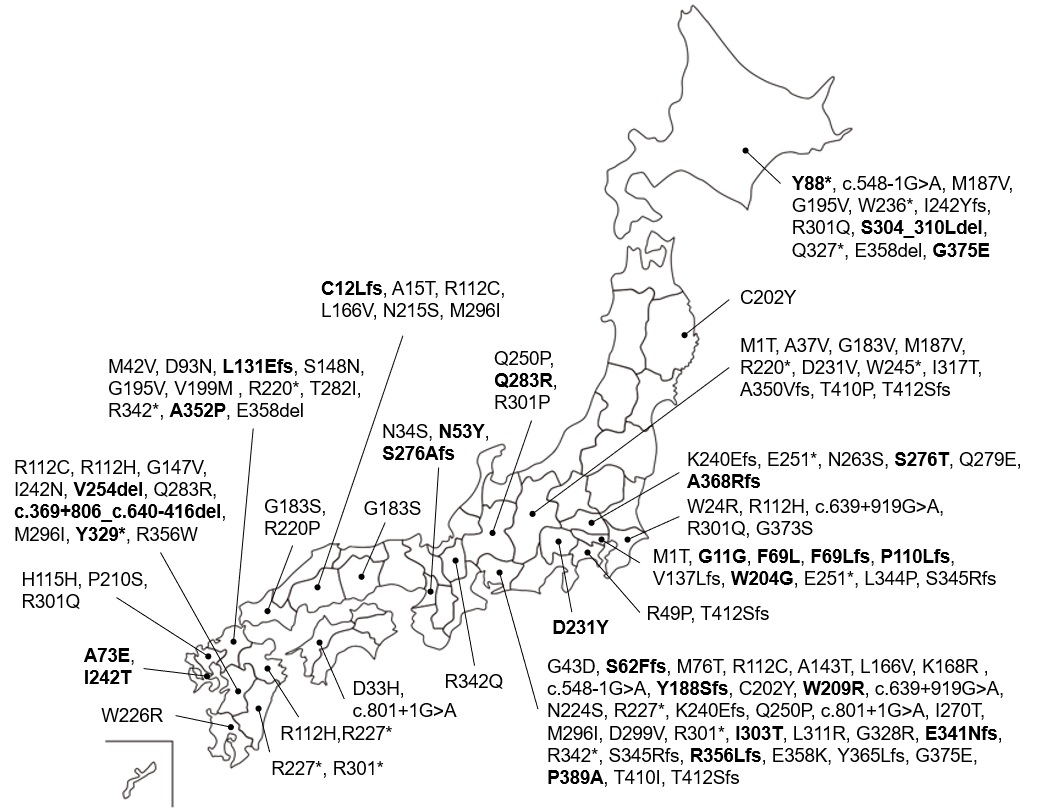
The variant spectra of GLA in Japanese patients have been reported [26, 27]. GLA gene analysis was performed for 207 Fabry patients [26]. The most common variant was c.888G>A/p.M296I (allele frequency: 5.8%, 12/207). The second most common variants were c.639+919G>A (4.3%, 9/207) and c.679C>T/p.R227* (4.3%, 9/207), followed by c.334C>T/p.R112C (3.9%, 8/207), c.335G>A/p.R112H (3.9%, 8/207), and c.902G>A/p.R301Q (3.9%, 8/207). In another study, 73 pathogenic variants were detected in 176 patients from 115 families [27] and the most common variant was c.334C>T/p.R112C (allele frequency: 2.65%). The second most common variant was c.888G>A/p.M296I (1.89%), followed by c.658C>T/p.R220* (1.52%), c.718_719delAA/p.K240Efs*8 (1.52%), and c.1025G>A/p.R342Q (1.52 %). The common variants identified in these studies, as well as those of the current study, overlap.
We previously reported the first large-scale NBS program for FD in the western region of Japan [11]. A total of 599,711 newborns were screened and 26 GLA variants, including 8 novel variants, were detected in 57 newborns from 54 families. Of the 26 variants, 10 were also detected in the current study and most of them were detected in patients from the western region of Japan (Figure S1).
In the current study, 4 pedigrees (4/68, 5.9%) were perceived as de novo mutations (Table S2). The frequency might be comparable with those of previous studies from Japan (6.8% (5/74); Kobayashi et al. [28]), Italy (2.8% (3/108); Romani et al. [29]; and 14.3% (2/14); Morrone et al. [30]), Spain (4.5% (1/22); Rodriguez-Mari et al. [31]), and the United Kingdom (6.3 % (1/16); Davies et al. [32]). A high frequency of de novo mutations has been reported in X-linked disorders, such as Duchenne muscular dystrophy (DMD) and hemophilia A (F8), and de novo mutations account for approximately one-third of the mutations in these two disorders [33, 34]. The size and structure of the gene and its position within the genome may contribute to the high frequency of de novo mutations. The high rate of de novo mutations in the two abovementioned diseases is thought to be related to the large size of the genes (DMD: 2,400 kb, F8: 186.9 kb). The presence of CpG dinucleotides also increases mutational frequency [35]. The relatively low frequency of de novo mutations in the GLA gene may be owing to its small size (10.2 kb) and relatively high CpG dinucleotide content; for example, GLA contains 19 CpG sites (1/68 bases) in the coding region compared to F8 which has 68 CpG sites (1/104 bases). Of the 19 potential mutation sites in GLA, 13 variants were identified in the current study, namely, c.146G>C, c.334C>T, c.335G>A, c.427G>A, c.658C>T, c.659G>C, c.679C>T, c.901C>T, c.902G>A, c.902G>C, c.1024C>T, c.1025G>A, and c.1066C>T. Twenty-three variants were reported as de novo mutational hotspots (Table S2). However, no particular sites responsible for these de novo mutations were identified.
A few case reports regarding the homozygous or compound heterozygous female Fabry patient have been reported [30, 36]. However, homozygous or compound heterozygous female Fabry patients were not identified in the current study, or our previous NBS study [11]. Generally, in female Fabry patients, only those with heterozygous mutations in the GLA gene are detected. The reason for this is not fully understood and should be further investigated. Interestingly, a male patient who possessed two GLA variants, c.70T>A /p.W24R and c.1255A>G/p.N419D, was detected in the current study (Table S1). Unfortunately, genetic information regarding his family was not available. Of 100 GLA variants, 70 were detected only in single pedigrees whereas 20 were identified in two pedigrees. Because bias was introduced in the distribution of variants in these pedigrees, it was difficult to discuss the correlation between genotype and phenotype, especially organ specific pathogenicity.
In the follow-up study of each patient, 21 of the 26 novel variants were indicated as pathogenic, namely c.97G>C/p.D33H, c.157A>T/p.N53Y, c.184dupT/p.S62Ffs*18, c.205T>C/p.F69L, c.207del/p.F69Lfs*52, c.264C>G/p.Y88*, c.329del/p.P110Lfs*11, c.386_389dupTGAA/p.L131Efs*9, c.440G>T/p.G147V, c.563delC/p.Y188Sfs*4, c.610T>G/p.W204G, c.691_693GAC>TAT/p.D231Y, c.825delC/p.S276Afs*6, c.827G>C/p.S276T, c.848A>G/p.Q283R, c.908T>C/p.I303T, c.987C>A/p.Y329*, c.1019delG/p.E341Nfs*57, c.1054G>C/p.A352P, c.1085_1088dupCTCG/p.Y365Lfs*11, and c.1100dupT/p.A368Rfs*7. There were also five variants identified which were not registered in ClinVar or Fabry-database.org, specifically c.725T>C/p.I242T, c.801+1G>A/p.L268Ifs*3, c.908_923del21/p.S304_310Ldel, c.1124G>A/p.G375E, and c.1165C>G/p.P389A. Patients who possess the above mentioned variants developed FD-related symptoms, and some had even passed away from a stroke or heart failure.
Even in a high-risk FD screening, individuals are given an uncertain diagnosis when the classic type symptoms of FD are absent and variants of unknown significance (VOUS) in the GLA gene are identified. This leads to a risk of misdiagnosis, inappropriate counseling, and extremely expensive treatment. Therefore, many researchers and clinicians strive to create a diagnostic algorithm for FD that can exclude these risks as much as possible [37]. In our high-risk screening, individuals presenting decreased activity (<cutoff levels) with known pathogenic variants, classic type signs or symptoms of FD and VOUS, or family history of FD and variants were definitely diagnosed with FD. However, in individuals presenting decreased activity (<cutoff levels) with VOUS and late onset signs or symptoms such as cryptogenic stroke, proteinuria, or LVH without classic type signs or symptoms, a definite diagnosis is difficult. Moreover, because the disease state in the late onset type is likely not improved by ERT [38], the therapeutic effect of ERT does not allow for the diagnosis of FD. Therefore, a temporary diagnosis of FD may be given for individuals with VOUS and late onset signs or symptoms to receive treatment including ERT. Blood Lyso-Gb3 assays and tissue diagnosis in a myocardial or/and renal biopsy may be sufficient for the definite diagnosis of FD [39]. Moreover, analysis using iPSC technology, such as Gb3 accumulation in iPSC-derived vascular endothelial cells, may also lead to a definite diagnosis [40].
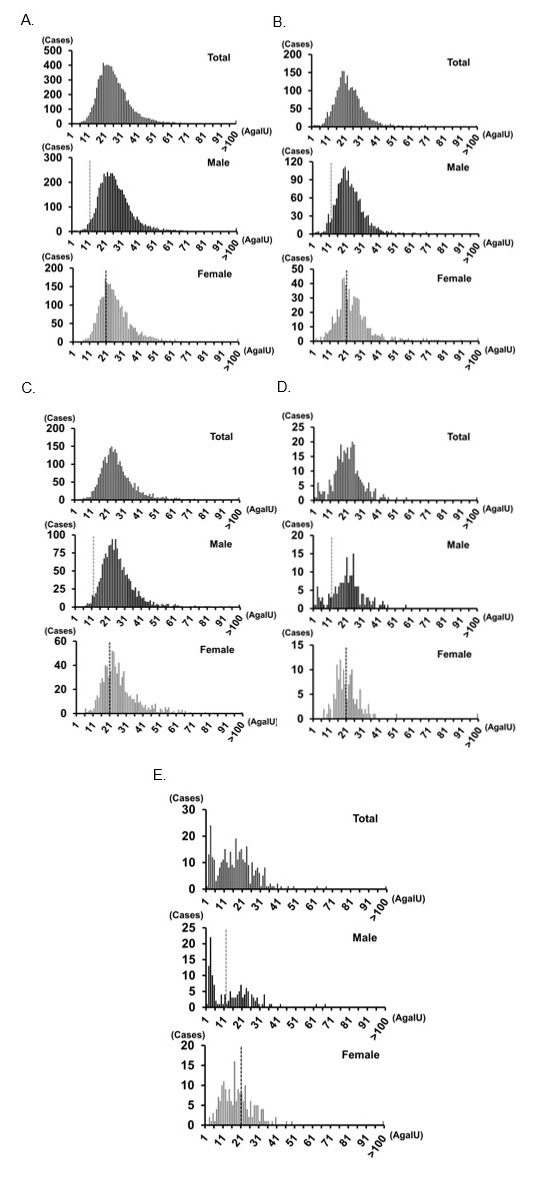
There is a potential for false negatives in a high-risk FD screening. Figures 2A, 2B, and 2C show the histograms of this high-risk screening for total, male, and female individuals, respectively, in Method I. The median α-Gal A activity was 24.47, 24.50, and 24.06 (AgalU) in total, male, and female individuals, respectively. A dotted line shows the cutoff value: <12 [AgalU] for males and <20 [AgalU] for females; 50% of median α-Gal A activity for males and 80% of median α-Gal A activity for females. It is known that heterozygous female patients have an almost normal range of α-Gal A activity, which causes false-negative results in screening studies. Linthorst et al. [41] demonstrated that 40% (16/40) of female patients with FD are not detected with the cutoff <50% of the normal control. Although we used a higher cutoff <80% of median α-Gal A activity, it is undeniable that there may be false-negative female patients with FD. Additional tests, such as blood Lyso-Gb3 assays [42], hotspot mutation screening [43], or even whole GLA gene sequencing, may improve the false-negative rate. Most patients with FD in Taiwan carry variants out of a pool of only 21 pathogenic mutations [43]. Therefore, in an area such as Taiwan, where the hotspot mutation can be detected, hotspot mutation screening is effective for female high-risk screening. In an area such as Japan, where the hot spot mutation cannot be detected and several variants are found, hotspot mutation screening is not effective. Whole GLA gene sequencing is difficult to be applied for all female patients included in the high-risk screening because of cost-balance issues. The assay of Lyso-Gb3 in dried blood spots (DBSs) is considered an effective and realistic option for female high-risk screening [42]. We will consider applying Lyso-Gb3 assay in our high-risk screenings in the future.
Figure 2D presents a histogram of α-Gal A activity in an NBS study in Method I [11]. The median α-Gal A activity in newborns was 42.58 (AgalU), which is approximately two times higher than that of current high-risk screening populations. FD is associated with a significantly reduced life expectancy compared to that of the general population [44]. Although the detailed mechanism for the low α-Gal A activity in adults is unknown, it may be associated with a premature ageing process through the dysfunction of blood vessels. Therefore, ageing and low α-Gal A activity are closely related. The cutoff values in the high-risk screening populations were 12 (AgalU) for males and 20 (AgalU) for females, which is representative of the cutoff values for the 0.5 percentile in the NBS population. This is because α-Gal A activity in adults is lower than that of newborns.
The current high-risk screening program identified individuals who are considered suitable candidates for migalastat treatment. Some patients were already receiving migalastat treatment. Moreover, gene therapy holds promise in the effective treatment of a wide variety of diseases, and the clinical trials for gene therapy for FD are ongoing in Canada and USA (https://fabrydiseasenews.com/gene-therapy-for-fabry-disease/). In the future, the development of new treatment methods for FD, other than ERT, is expected.
{kind=link}
{kind=link}