Our initial studies toward this reaction explored the exchange of 13C-benzoyl chloride with p-anisoyl chloride. As sterically encumbered ligands have been noted to favor acid chloride reductive elimination,22,27 various catalysts incorporating large phosphine ligands were probed in this chemistry (Table S1). These show that palladium catalysis with the bidentate ligand Xantphos can mediate the 13C-exchange between these aromatic acid chlorides at temperatures as low as 50 oC (Fig. 2a). Unfortunately, when exchange was examined with an alkyl acid chloride, a significant amount of alkene was invariably observed, arising from the ability of these palladium systems to also undergo rapid β-hydride elimination. Similar results were noted with all ligands probed (Table S2).
In considering approaches to induce reversible CO insertion and disfavour β-hydride elimination, we questioned if a more productive reaction may be obtained by moving to nickel systems. Nickel catalysts are not commonly employed in carbonylation chemistry due in large part to their ability to bind more tightly to carbon monoxide than palladium thereby inhibiting catalysis.28 However, there is no extraneous carbon monoxide in this exchange reaction, and we postulated that the association of CO to nickel may instead inhibit its loss, and also block the formation of empty coordination sites classically required for alkene de-insertion. In addition, nickel is known to be much less susceptible to β-hydride elimination29 and can mediate decarbonylative transformations,24,25 suggesting that at least two of the needed steps in catalysis are viable. As was hoped, the use of nickel catalysts with various phosphines or nitrogen ligands completely inhibits β-hydride elimination, although only low levels of 13CO exchange were observed (Fig. 2b, Table S3). Since electron rich nickel complexes often undergo sluggish reductive elimination reactions,29 the reaction was next probed without donor ligands. These show that the use of simple Ni(COD)2 in concert with a chloride source to the stabilize Ni(0) allowed a rapid reaction at just ambient temperature to form the labeled alkyl acid 1b-13C with high levels of isotope exchange and no detectable amount of β-hydride elimination.
Ni(COD)2 is a simple, commercially available catalyst, but is also air sensitive. In considering complementary catalyst designs, another possibility for this reaction would be to exploit CO itself as a ligand. Nickel carbonyl complexes of the form LNi(CO)3 (L = phosphine) are air stable, and the π-acidic carbon monoxide ligand could create an electron-poor nickel prone to reductive elimination. A range of (R3P)Ni(CO)3 catalysts were tested and found to mediate rapid 13C exchange in the presence of a chloride salt (Fig. 2b, Table S3). In particular, the sterically encumbered phosphite coordinated catalyst (CO)3Ni(P(O-2,4-tBuC6H3)3) (L1Ni(CO)3), is even more active than Ni(COD)2, and led to near complete exchange within 4 h. Of note, the air stable catalyst can be handled on the bench for reaction set up with only slightly diminished exchange (Table S3). In addition to using acid chloride as reagents, the reaction can be conveniently coupled with in situ chlorination of the carboxylic acids with TMCE (1-chloro-N,N,2-trimethyl-1-propenylamine) and the solid, easily handled and long lived 13C donor or 14C donor, 4-phenyl benzoic acid.30,31 Subsequent nickel exchange can offer a straightforward route to incorporate carbon-label into the parent carboxylic acid (Fig. 2b).
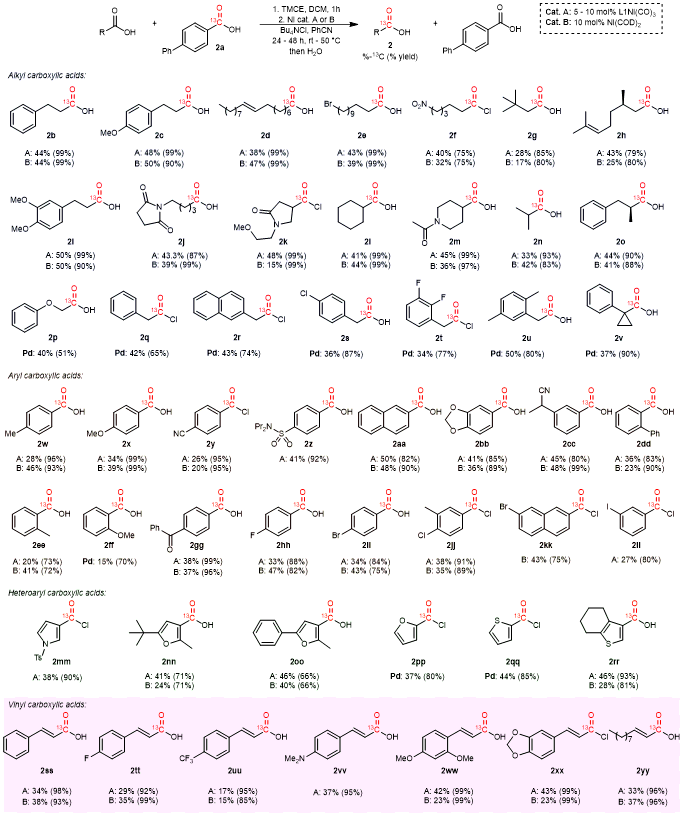
With two complimentary catalysts systems in hand, the commercially available Ni(COD)2 or the air stable L1Ni(CO)3, we next explored the generality of this approach to carbon labeling. As was hoped, a diverse array of alkyl carboxylic acids can be employed in this reaction (Table 1). Primary alkyl substrates such as hydrocinnamic acid derivatives (2b, 2c, 2i) undergo efficient exchange with both catalysts, as do the more sterically encumbered β-branched 2g or 2h. High levels of exchange are noted with substrates incorporating possibly sensitive alkene (2d), alkyl bromide (2e), imide (2j) and even nitro (2f) functional groups. Secondary alkyl carboxylic acids are also viable in the reaction (2k-o), including those with lactam (2k) and amide (2m) functional groups. Similar results are observed when performing the reaction on larger scale (e.g., 2j). Coordinating functionalities in some substrates slow exchange with Ni(COD)2 (e.g., 2h, 2k), potentially due to their association to the undercoordinated nickel, but these undergo excellent exchange with L1Ni(CO)3. In addition to exchange, no racemization occurs in reactions with chiral substrates (2h, 2o), including those with α-stereogenic centers. The latter contrasts to recent metal-free platforms for isotopic labeling, and offers a synthetic route to label chiral carboxylic acids without the need for subsequent product resolution.
The approach can also be applied to label aromatic carboxylic acids. Simple (2aa), electron rich (2w, 2x, 2bb, 2cc), and electron poor aryl carboxylic acid (2y, 2z, 2gg) are viable CO exchange substrates, as are ortho- substituted substrates (2dd, 2ee). Catalysis is similarly efficient with acid chlorides containing potentially reactive aryl halides (2hh-2ll) with L1Ni(CO)3. 13C exchange occurs with coordinating heterocyclic carboxylic acids at slightly elevated temperatures (50°C), including pyrroles (2mm), furans (2nn, 2oo) and thiophenes (2rr). α,β-Unsaturated carboxylic acids are similarly viable in this exchange, including electron neutral (2ss), electron rich (2vv, 2ww, 2xx), and electron poor (2tt, 2uu) cinnamic acid derivatives, or acyclic (2xx, 2yy) substrates. In general, these data suggest that both catalysts are viable for 13C exchange, where Ni(COD)2 is a slightly more active catalyst, but more easily subject to deactivation by associating or sensitive functionalities than the coordinatively saturated L1Ni(CO)3. In instances where the exchange is poor with the nickel catalysts, the palladium system was found to offer complementary reactivity. Examples include benzoic acids with coordinating groups on the ortho-position (2ff), 2-heteroaryl substrates (2pp, 2qq), or benzylic carboxylic acids (2q-v), including the tertiary, cyclopropyl-containing 2v. Together, this exchange reaction offers access to labeled products with the broadest range of reactivity of which we are aware (alkyl, benzyl, aryl, heteroaryl or vinyl carboxylic acids), and does so using a single, easy to handle carbon-donor without pressing conditions or specialized setups.
We next explored how the nickel catalyzed exchange proceeds. Monitoring catalysis by in situ 31P NMR analysis shows the slow loss of L1Ni(CO)3 throughout the reaction and growth of free phosphite (Fig. 3a). In addition, in situ 13C NMR analysis reveals the slow incorporation of 13C label into L1Ni(CO)3 (δ 194.1 ppm). The build-up of free phosphite, together with the high catalytic activity with this weak donor ligand and added chloride, suggests that the displacement of the phosphite by chloride may be important for catalysis. Consistent with this postulate, control experiments show the phosphite free [(CO)3NiCl−][Bu4N+] is also a potent catalyst for the reaction (Fig. 3b), albeit one that is unstable and must be generated in situ,33 while the addition of free phosphite to catalysis with L1Ni(CO)3 inhibits the reaction (Fig. 3c). In order to probe how acid chloride activation occurs with this nickel system, catalysis was performed in the presence of TEMPO, which completely inhibits exchange and forms a catalytic amount of the two acyl-TEMPO adducts (Fig. 3d), consistent with a radical oxidative addition mechanism. We observe similar radical trapping products the Ni(COD)2/Bu4NCl system (Fig. 3d). Nevertheless, the lack of racemization of α-chiral carboxylic acids (Table 2, 2o) shows that CO loss for exchange does not occur from the radical itself, but presumably instead does so on nickel. The reaction can be performed in an open flask, also suggesting minimal free CO formation (Fig. 3b).
Overall, these data are consistent with the mechanism shown in Fig. 3e, wherein single electron transfer from Ni(0) (e.g., I) to acid chloride forms an acyl radical that can undergo rapid recombination to form the nickel-acyl complex II. Supporting these steps, performing catalysis with stoichiometric Ni(COD)2 leads to the radical coupling ketones and diketone as the now major products (Figure S1). Interestingly, the same reaction with stoichiometric L1Ni(CO)3 proceeds in a similar fashion to that with catalytic nickel, suggesting the slow release of phosphite may keep acyl radical concentrations lower during the reaction (Figure S2). Once II is generated, reversible CO de-insertion/insertion would allow the incorporation of label into the acyl ligand on a single nickel catalyst, and without the need for either exchange between two metals, as has been previously suggested for palladium in dynamic reactions, 20,21 or the release of free CO. The electron deficient, CO associated Ni(II) intermediate presumably undergoes rapid acid chloride elimination to generate the Ni(0) complex as the only observable catalyst resting state (Fig. 3a). The presence of multiple CO ligands on nickel likely facilitates rapid CO insertion to inhibit β-hydride elimination and allow for selective generation of acid chloride products with an array of substrates.
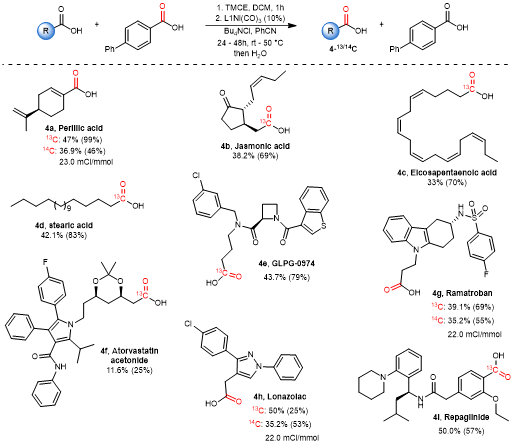
Finally, we have probed the applicability of this system to the carbon-isotopic labeling of biologically relevant molecules. As examples, carboxylic acid-containing natural products can be directly labeled with this platform (Table 2). These include the α,β-unsaturated carboxylic acid of the terpenoid perillic acid (4a), jasmonic acid (4b) and the polyene containing fatty acid eicosapentaenoic acid (4c), each of which affords good levels of isotopic incorporation and with no racemization. Pharmaceuticals and pharmaceutically relevant compounds are also viable substrates in this exchange. Representative examples here are GLPG-0974 (4e), a neutrophil-driven inflammation attenuating agent, which undergoes near quantitative exchange despite the presence of the azetidine, thianaphthene and amide functionalities, or the benzylic and functionalized aromatic carboxylic acids of the NSAID lonazolac (4h) and the antidiabetic drug repaglinide (4i) with palladium catalysis. Similar exchange is noted with the alkyl carboxylic acids in ramatroban (4g), an antagonist for thromboxane receptors and treatment of coronary artery disease, and the functionalized carboxylic acid in atorvastatin acetonide (4f). We have also performed radiolabeling of perillic acid (4a), ramatroban (4g) and lonazolac (4h) with 14C-4-biphenylcarboxylic acid as the isotope donor. All of these reactions afforded high levels of 14C specific activity, and highlight the ability of the protocol to readily incorporate carbon-14 label into pharmaceuticals.
{kind=link}
{kind=link}