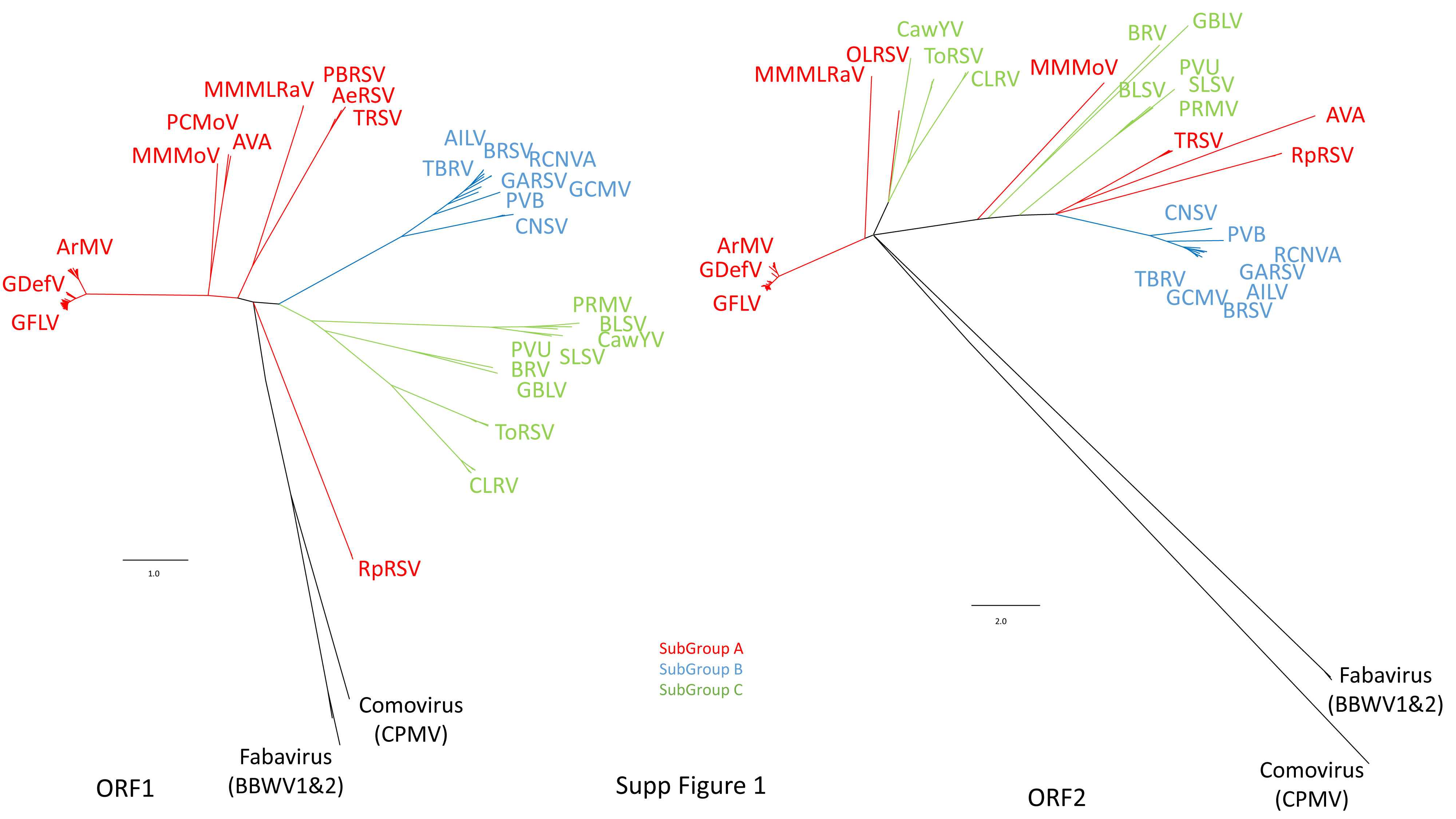
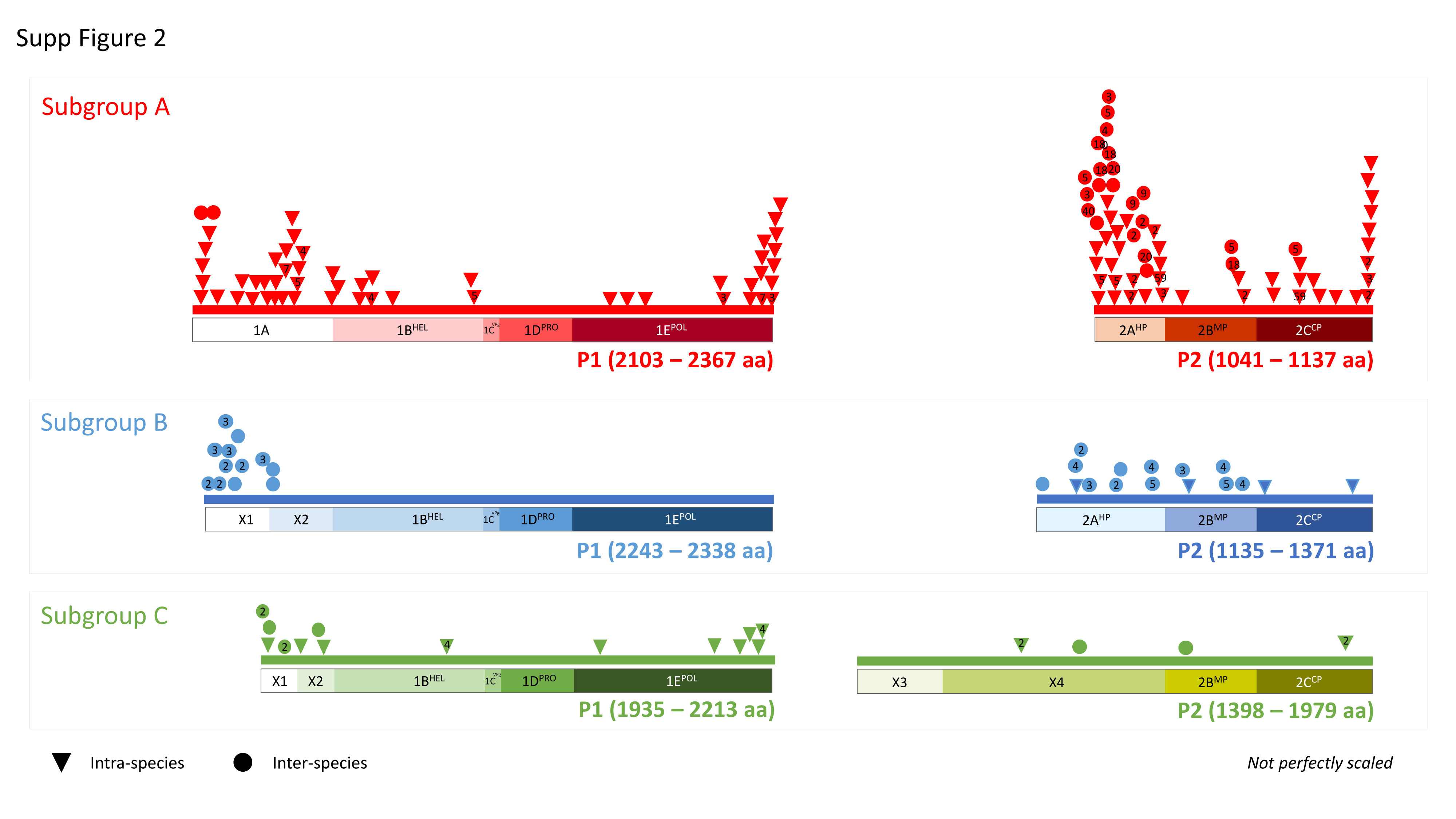
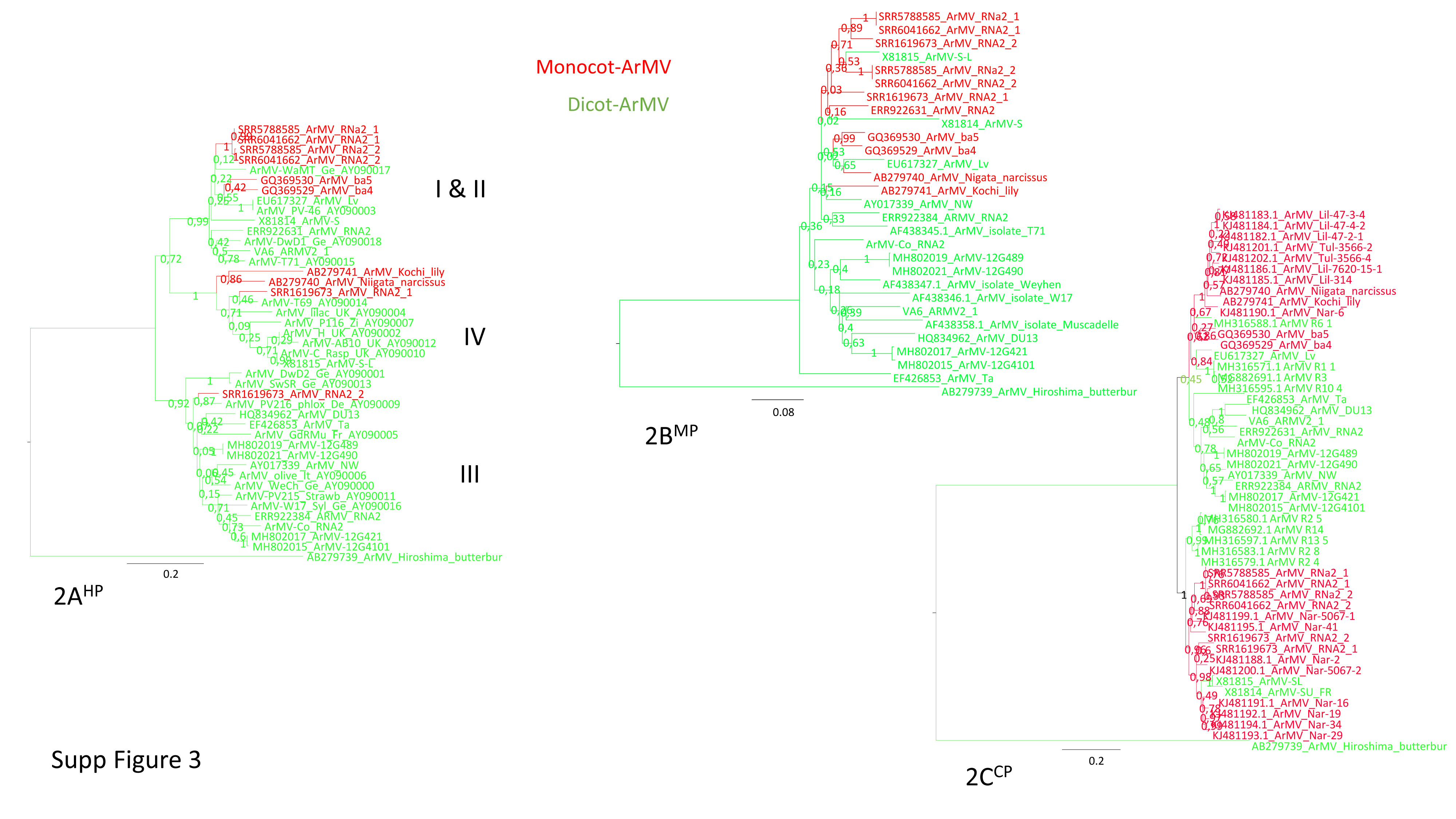
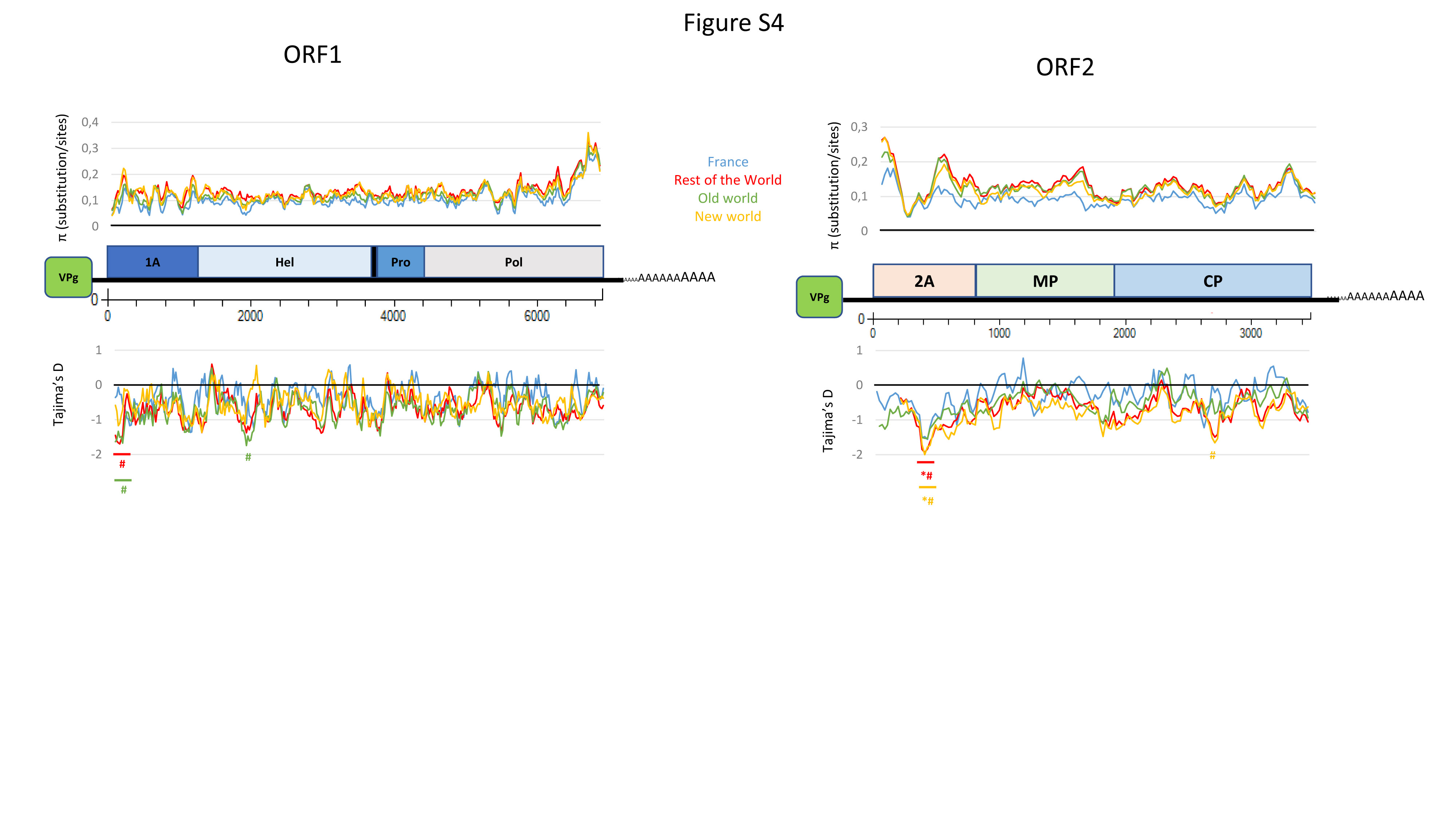
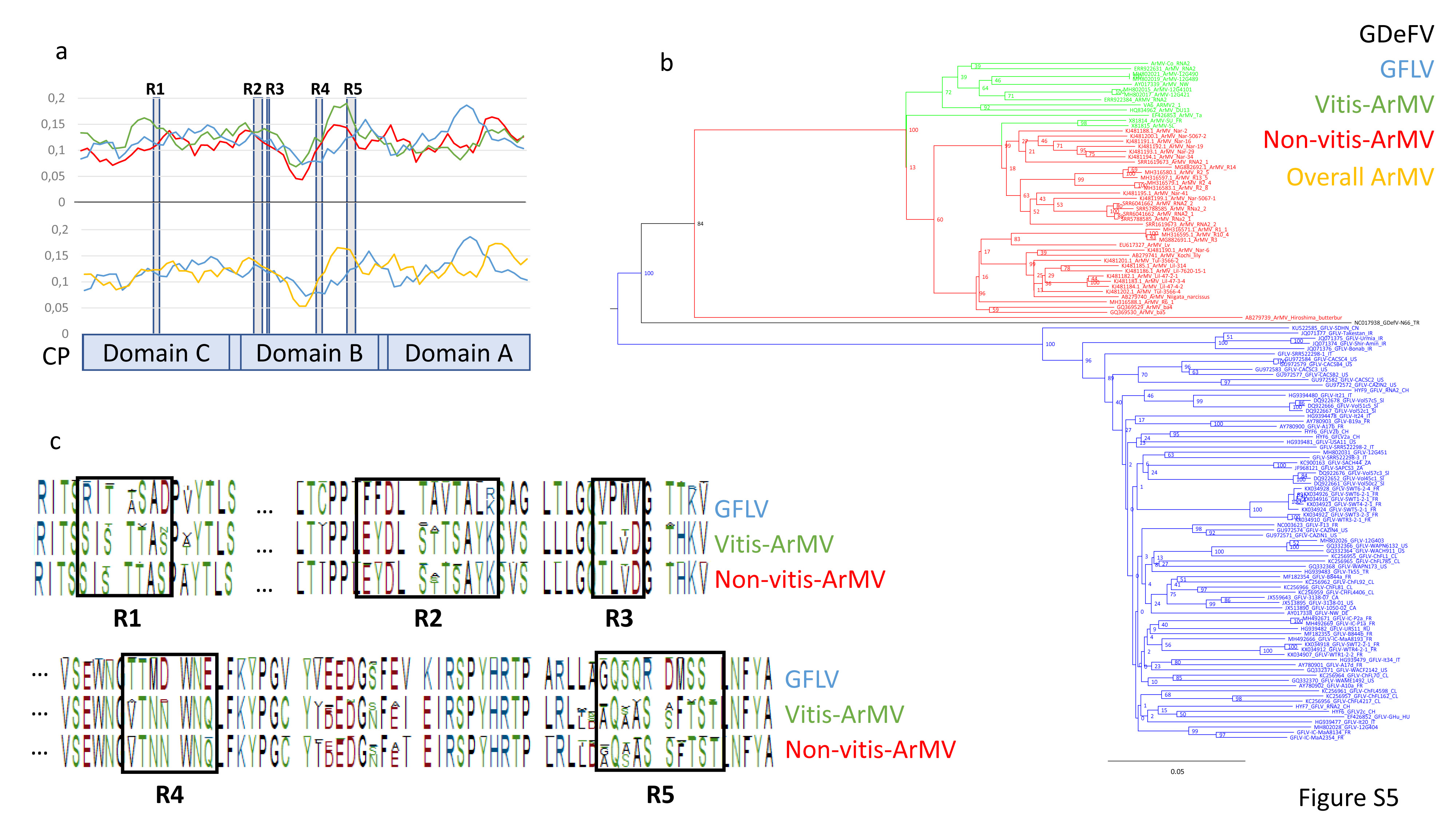
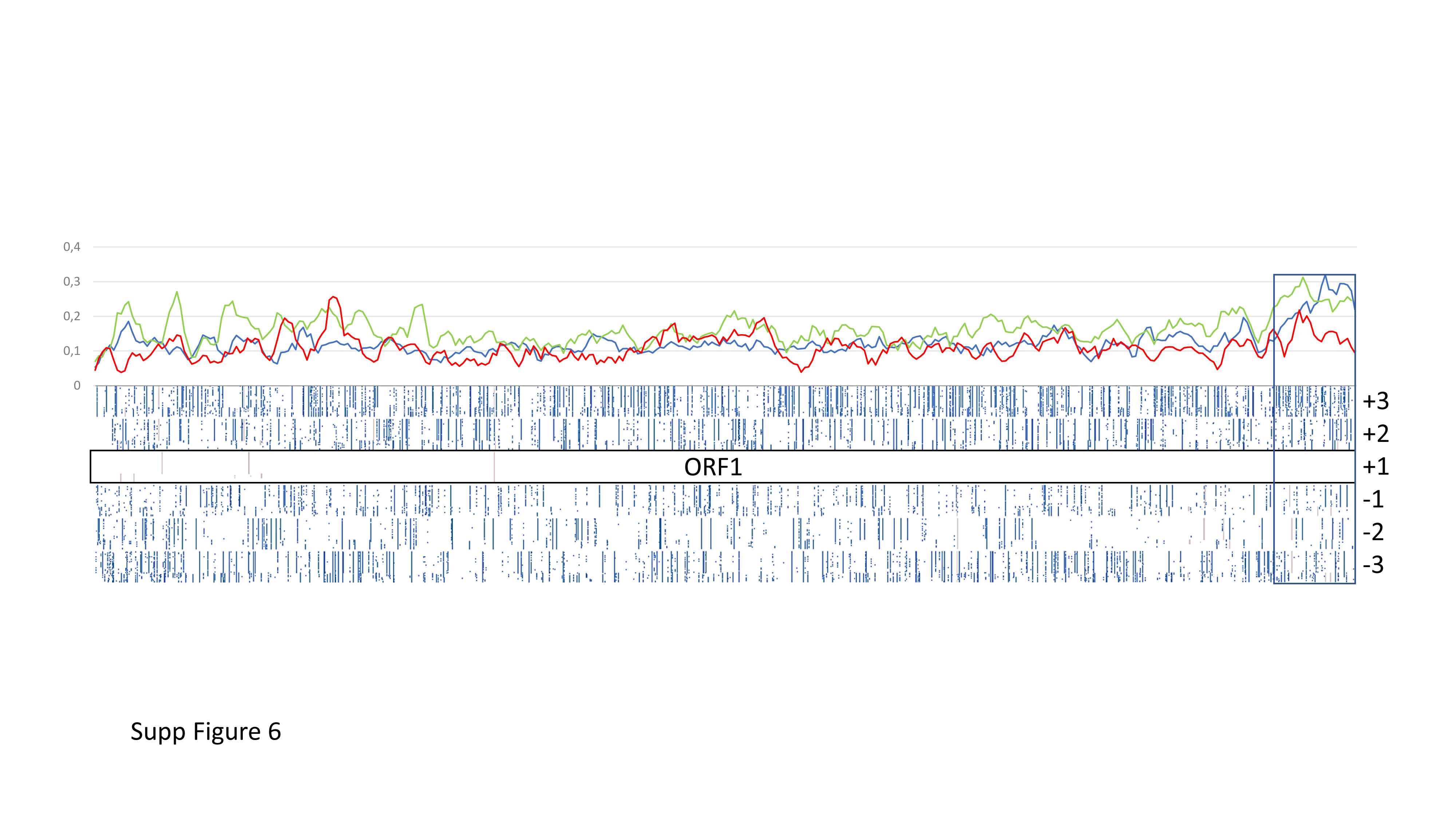
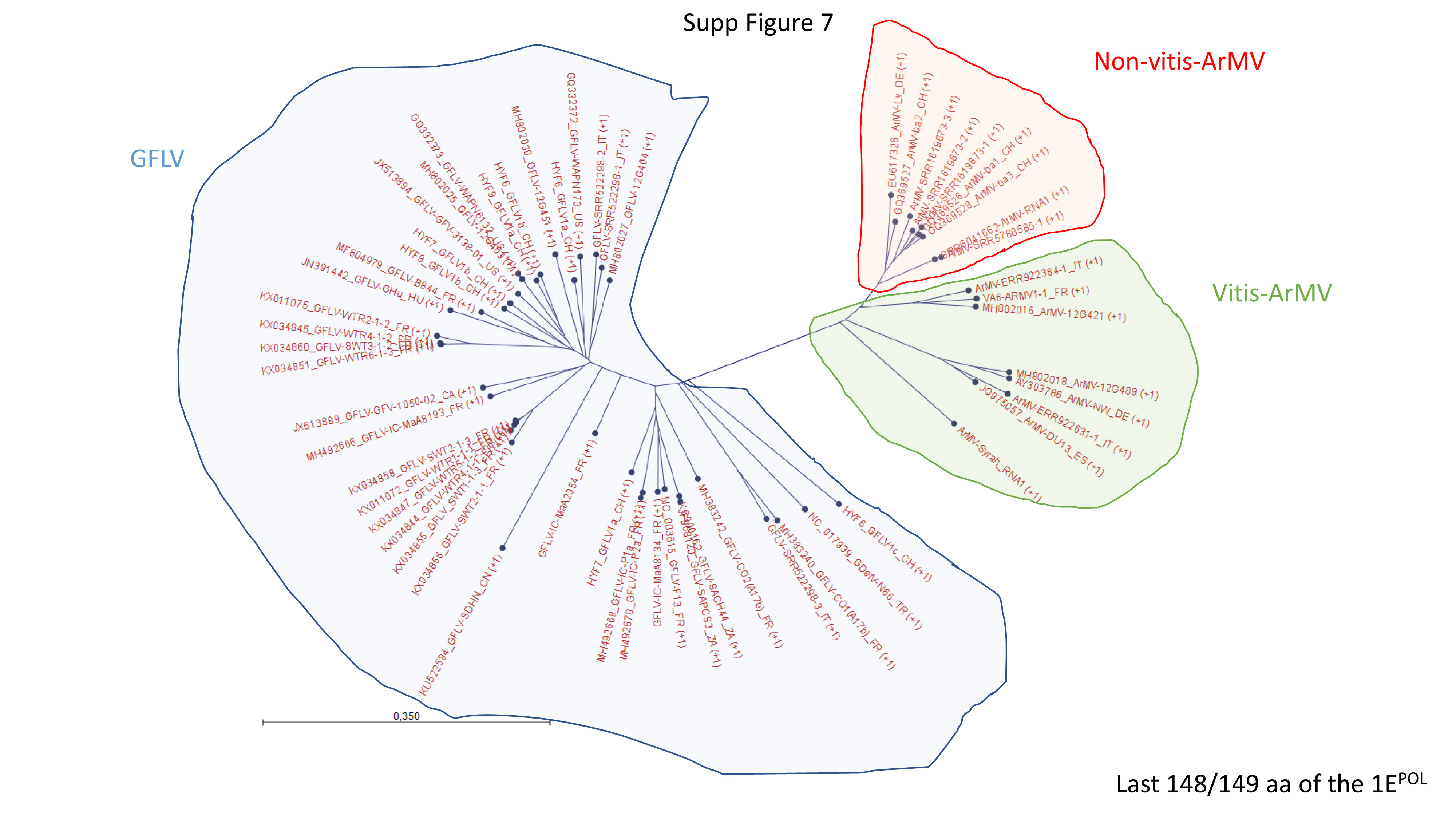
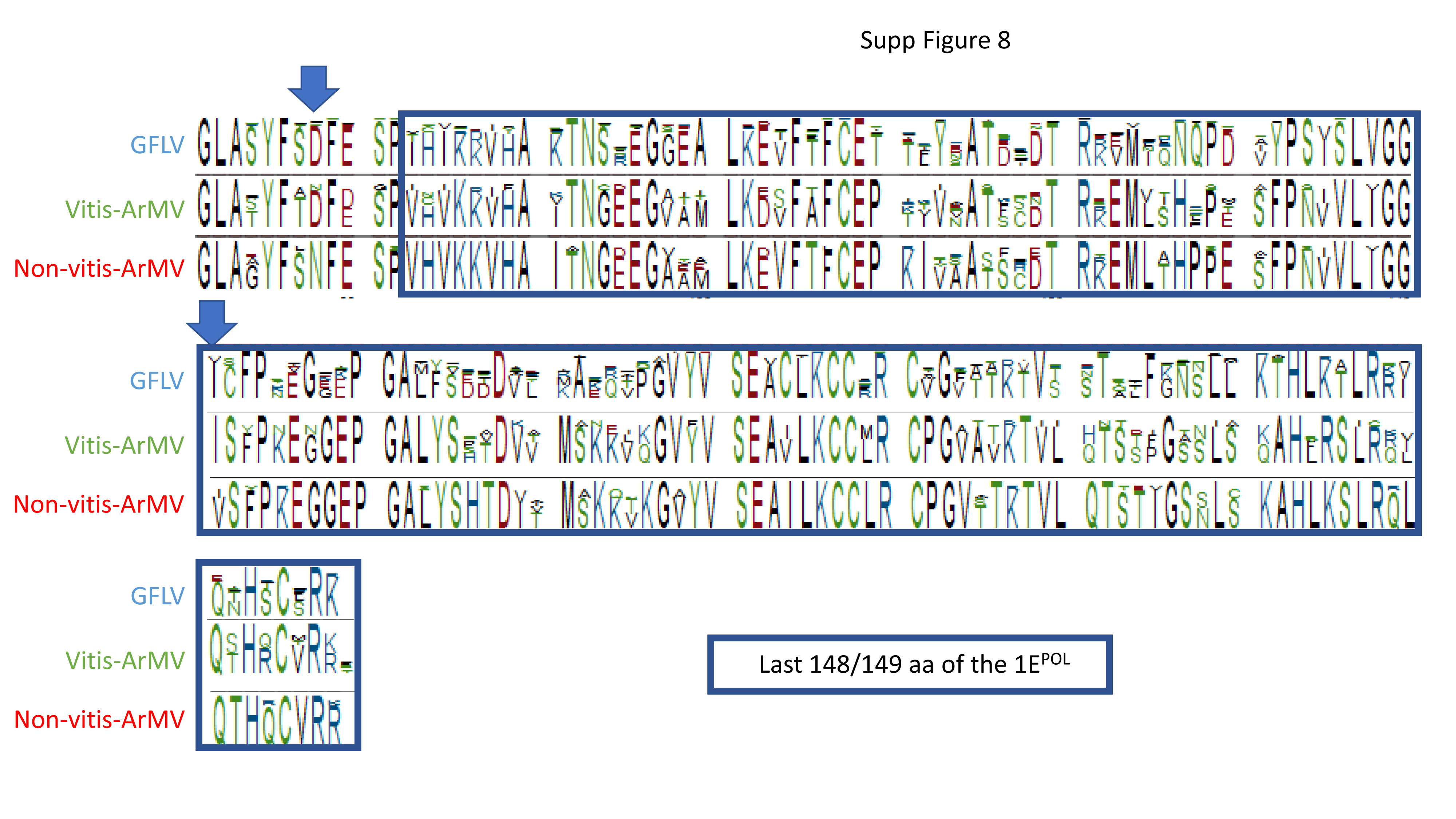
Datamining and metagenomic analyses of 277 open reading frame sequences of bipartite RNA viruses and variants in the genus Nepovirus documented how delicate it can be to unequivocally identify species, in particular subgroup A and C species, based on some of the currently adopted taxonomic demarcation criteria. It suggests a possible need for their amendment to accommodate pangenome information. In addition, we revealed a host-dependent structure of arabis mosaic virus (ArMV) populations at a cladistic level and confirmed a phylogeographic structure of grapevine fanleaf virus (GFLV) populations. We also identified new putative recombinant events for species of subgroups A, B and C. The evolutionary specificity of some capsid regions of ArMV and GFLV that were previously described and biologically validated as vector determinant was circumscribed in silico. Furthermore, a C-terminal segment of the RNA-dependent RNA polymerase of subgroup A species was predicted as a putative host range determinant based on statistically supported higher π values for GFLV and ArMV isolates infecting Vitis spp. compared to non-Vitis infecting ArMV isolates. This study illustrated how sequence information obtained via high throughput sequencing can increase our understanding of mechanisms that modulate virus diversity and evolution and create new opportunities for advancing studies on the biology of economically important plant viruses.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}