Identification of DEGs
A total of 3520, 424, and 1675 DEGs were identified from GSE41657, GSE44861, and GSE74602 profiles, respectively. Two hundred twenty-three genes were screened out in all three datasets and were selected for further analysis (Fig. 1). There are 184 up-regulated genes (Fig. 1A) and 39 downregulated genes (Fig. 1B) in CRC tissues compared to adjacent tissues.
GO and KEGG Pathway Enrichment Analyses
To explore the internal relations, we uploaded the differently expressed genes to the online website DAVID to identify GO Terms and KEGG pathways and classified them into three functional categories: biological process (BP), cellular component (CC), and molecular function (MF).
In the BP category, the up-regulated gene enrichment GO terms were mainly proteolysis, transport, ion transmembrane transport, response to drug and bicarbonate transport, while the downregulated gene enrichment GO terms were mainly positive regulation of neutrophil chemotaxis, chemokine-mediated signaling pathway, canonical Wnt signaling pathway, response to a drug, positive regulation of protein oligomerization, response to lipopolysaccharide and regulation of cell proliferation (Fig. 2B). Besides, in the CC category, the differently expressed gene enrichment GO terms were mainly extracellular exosome, extracellular space, apical plasma membrane, perinuclear region of cytoplasm, and integral component of the plasma membrane, while the downregulated gene enrichment GO terms were mainly extracellular space and extracellular matrix. Moreover, in the MF category, the differently expressed gene enrichment GO terms were mainly zinc ion binding, calcium ion binding, chloride channel activity, hormone activity, and oxidoreductase activity. In contrast, the downregulated gene enrichment GO terms were mainly calcium ion binding, CXCR chemokine receptor binding, chemokine activity, and metalloendopeptidase activity.
As for the KEGG pathway, the most significant results were eight up-regulated genes (MT2A, MT1M, MT1F, MT1G, MT1H, MT1X, SLC26A3, MT1E) involved in Mineral absorption (Fig. 2C) and five downregulated genes (CXCL8, CCND1, MMP1, MYC, MET) involved in pathways in cancer (Fig. 2D).
Construction of PPI network
The PPI network of the differently expressed genes was constructed with 164 nodes and 371 edges by using the STRING database and Cytoscape 3.9.1 software (Fig. 3). Scores of PPI network nodes were calculated by Cytoscape 3.9.1 built-in plugin cytoHubba. Then, the top ten genes, including MYC, CCND1, MMP7, TIMP1, MMP1, MMP3, CXCL2, CXCL1, CXCL8, and CXCL12, were identified as hub genes (Table 1).
Table 1
Hub genes screened from PPI.
|
Gene name
|
MCODE Score
|
Degree
|
|
MYC
|
7
|
21.0
|
|
CCND1
|
7
|
20.0
|
|
MMP7
|
6.377777778
|
12.0
|
|
TIMP1
|
6.377777778
|
15.0
|
|
MMP1
|
6.377777778
|
10.0
|
|
MMP3
|
6.377777778
|
11.0
|
|
CXCL2
|
5.303030303
|
12.0
|
|
CXCL1
|
5.303030303
|
17.0
|
|
CXCL8
|
4.846153846
|
20.0
|
|
CXCL12
|
4.846153846
|
17.0
|
mRNA Expression and Survival Analysis.
GEPIA2 database was used to analyze the expression levels of the ten hub genes. Compared with normal samples, colorectal cancer samples had higher expression of MYC, CCND1, MMP7, TIMP1, MMP1, MMP3, CXCL2, CXCL1, and CXCL8. The expression of CXCL12 was lower in colorectal cancer samples than in normal samples (Fig. 4).
GEPIA2 was used to predict the prognostic value of the ten hub genes, perform survival analysis and plot a Kaplan-Meier curve. Our results showed that high TIMP1 expression was associated with the worse overall survival of colorectal cancer patients (p < 0.05) for all colorectal cancer cases. In addition, there was no significant correlation between the differential expression of other Hub genes and the prognosis of patients (Fig. 5).
Validation of potential biomarkers in CRC Tissues
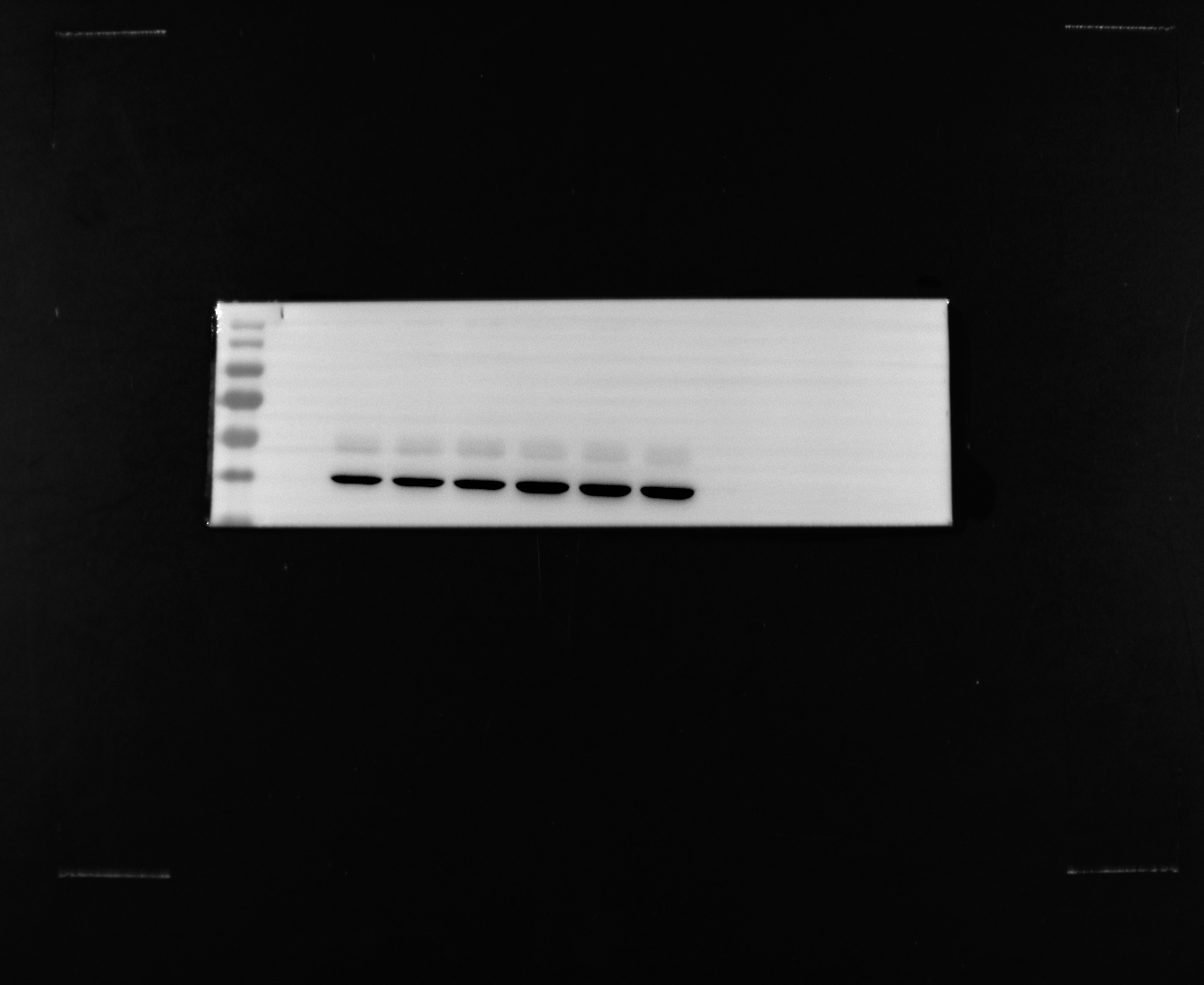
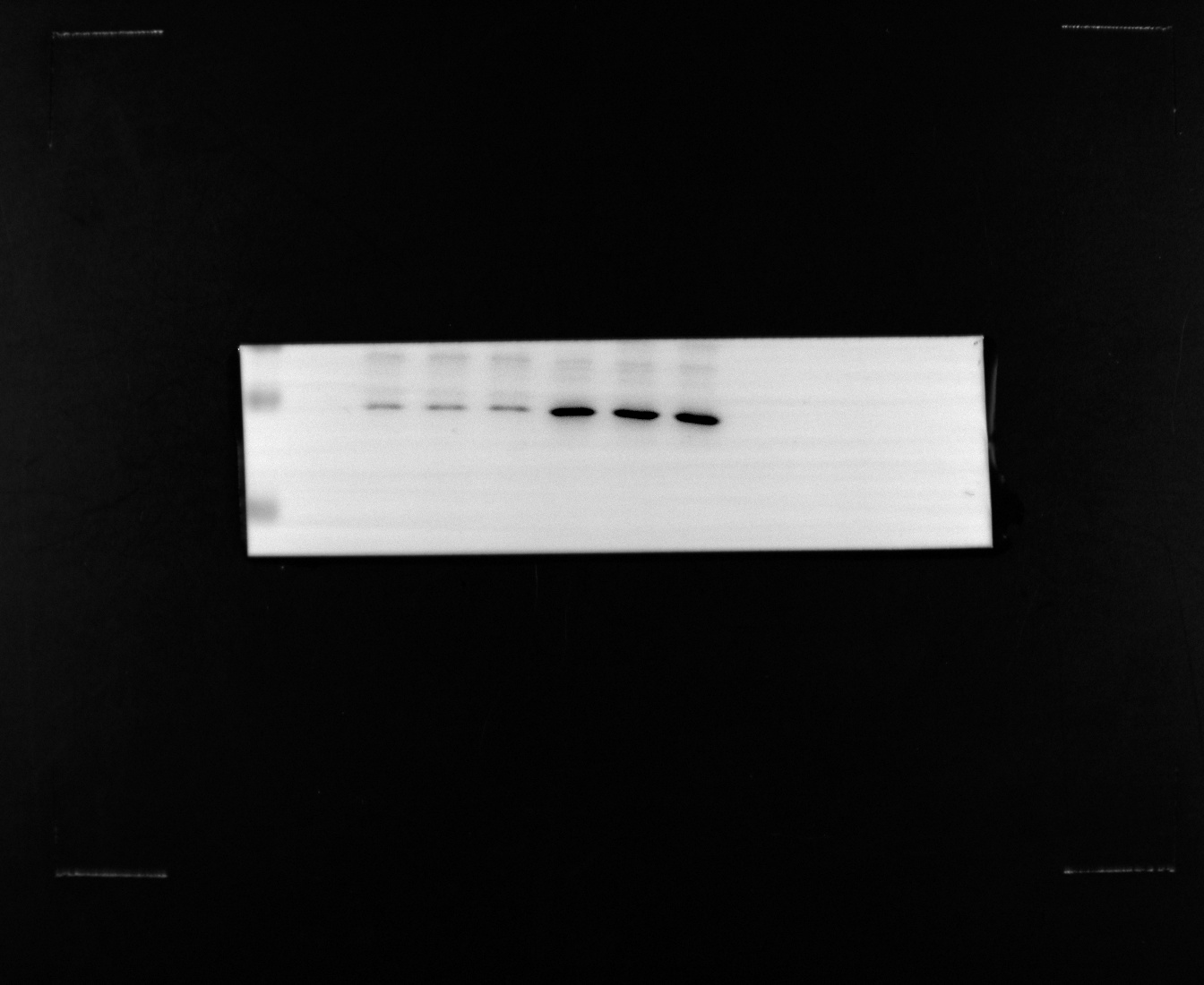
To further verify the results of the above omics data analysis, we collected tissue samples from colorectal cancer patients in the First Affiliated Hospital of Zhejiang Chinese Medicine University. Protein and nucleic acid samples are obtained after tissue samples are lysed at low temperatures. RT-qPCR and western blot verified the mRNA and protein expression trends of the potential biomarker TIMP1. The results showed that TIMP1 was significantly overexpressed in CRC tissues compared with the control group at DNA transcription and protein expression levels (Fig. 6A, B, and C).
{kind=link}
{kind=link}