Clinical specimens
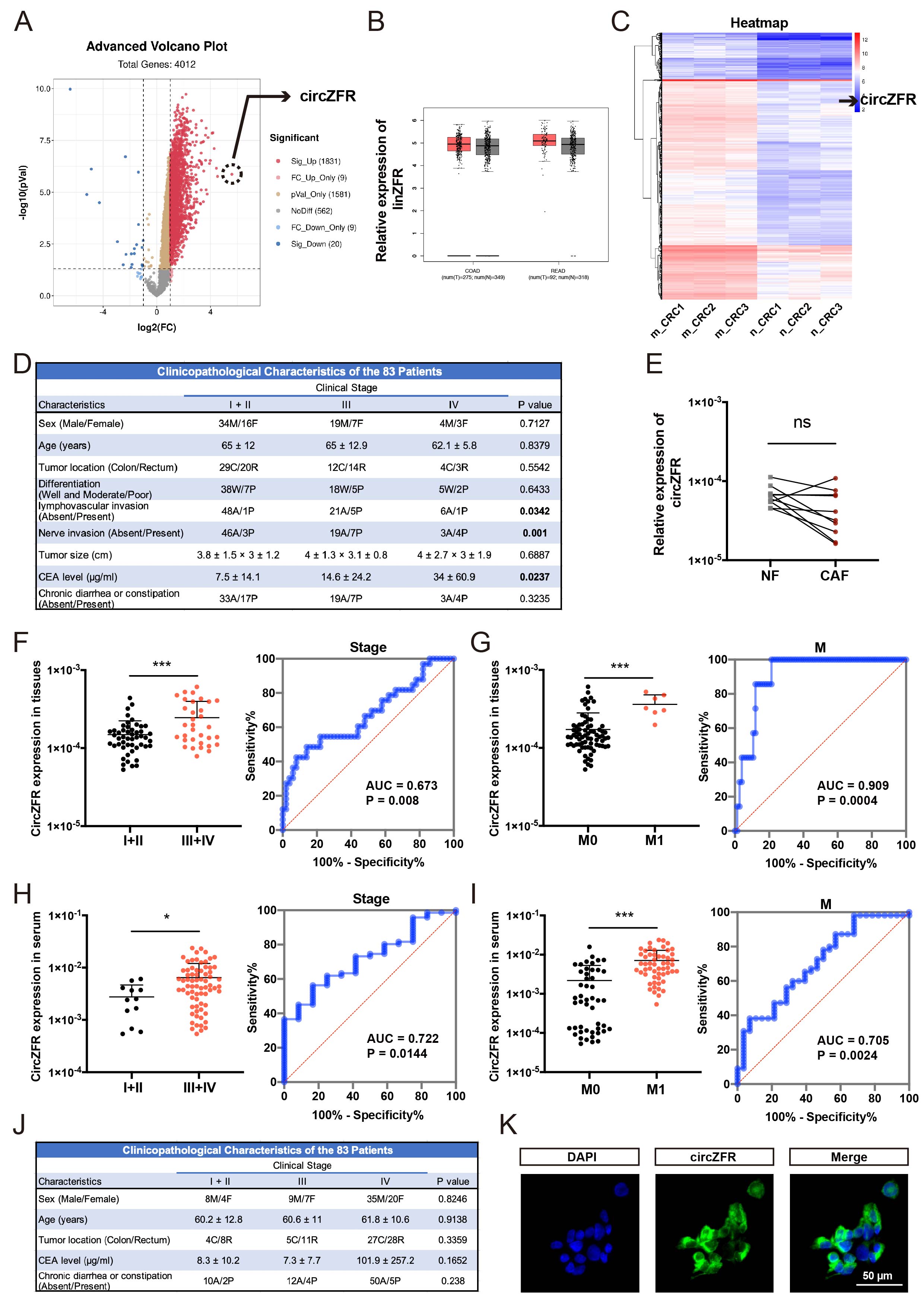
All 83 pairs of CRC tissues and corresponding adjacent normal tissues were obtained from CRC patients during operation between March 2019 and March 2021 at the Sir Run Run Shaw Hospital, Hangzhou, China. All 83 peripheral serum samples were collected from patients with CRC before operation between November 2018 and December 2020 at the Sir Run Run Shaw Hospital, Hangzhou, China. The inclusion criteria of patients were as follows: primary CRC with clear pathological diagnosis, no preoperative treatment including systemic chemotherapy and local radiotherapy, and complete clinicopathological records. Nevertheless, many patients that provided serum samples were diagnosed at advanced stages and had no indication for surgery. Therefore, clinicopathological features including tumor differentiation, lymphovascular or nerve invasion, and tumor size were unable to collected. Meanwhile, another 83 serum samples were also collected from sex- and age- matched healthy donors who underwent physical examination in the Sir Run Run Shaw Hospital during the same time period. The exclusion criteria of healthy population were as follows: diabetes, chronic systemic disease and obesity. For the construction of tissue microarray (TMA), 180 pairs of CRC and adjacent normal tissues were collected from patients with complete follow-up data during operation between October 2011 and August 2012 at the Sir Run Run Shaw Hospital.
The CRC stage was defined following the rules of the 8th edition of the American Joint Committee on Cancer (AJCC) tumor-node-metastasis (TNM) staging system. Among the 83 cancer serum samples, 3 stage Ⅱ and 3 stage Ⅳ samples were used for ceRNA microarray. All tissue and serum samples were collected with informed consent of patients and healthy donors and immediately stored at -80°C until use. This study was approved and monitored by the Ethics Committee of the Sir Run Run Shaw Hospital, Zhejiang University.
Cell culture and treatment
A panel of CRC cell lines, including HCE8693, SW480, SW620, DLD-1, RKO, Colo320 and HCT116, and human embryonic kidney cells (HEK-293T, referred to as 293T hereafter) were cultured in Dulbecco’s modified essential medium (DMEM) or RPML-1640 medium (Gibco BRL, Rockville, MD) with 100 µg/mL streptomycin, 100 U/mL penicillin and 10% fetal bovine serum (FBS; Gibco, NY, USA) and incubated at 37°C in a 5% humidified CO2 atmosphere. Cells were treated with cycloheximide (CHX; Sigma, MO, USA) at 50 µg/mL for the indicated time period, MG132 or Chloroquine (Selleck Chemicals, Shanghai, China) at 20 µM or 10µM, respectively, for 12 h, PR619 (Selleck Chemicals, Shanghai, China) at 20 µM for 48–72 h.
Protocol for the isolation of cancer-associated fibroblasts (CAFs) and normal fibroblasts (NFs) derived from fresh CRC tissues and paired normal tissues was fully described in our published article[22].
Competing endogenous RNA (ceRNA) microarray
The ceRNA microarray were conducted according to the protocol of Agilent technologies at LC Sciences Corporation (Hangzhou, China). Total RNA was isolated and purified by using RNA Isolation Kit (Norgen Biotek, Thorold, Canada) and RNase Mini Kit (Qiagen, Duesseldorf, Germany). The RIN number was then checked to inspect RNA integration by an Agilent Bioanalyzer 2100 (Santa Clara, CA, USA). Next, exosomal RNA samples of m-CRCs and n-CRCs were used for the LC Human ceRNA Array V1.0 to generate biotinylated cRNA targets which hybridized with the sliders. After hybridization, sliders were scanned on the Agilent Microarray Scanner (Santa Clara, CA, USA). Finally, data were extracted with Feature Extraction software (Santa Clara, CA, USA) and normalized by Quantile algorithm.
RNA extraction and quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted from tissues, serum or cells using the TRIzol reagent (Invitrogen, MA, USA), and RNA concentration was measured by Nanodrop 2000. RNA was treated with 3 U/µg RNase R (Epicenter Technologies, Madison, WI, USA) for 15 min at 37°C, or 5 µg/mL actinomycin D (AAT Bioquest, CA, USA) and collected at the indicated time points. Then, RT-qPCR were performed using an Evo M-MLV RT Premix kit (Accurate Biotechnology (Hunan) Co., Ltd) and SYBR FAST Universal qPCR kit (KAPA, Wilmington, USA) as described in our published article[22].
Exosome isolation and characterization
Exosomes were isolated from serum and culture medium of CRC cells following the standard centrifugation steps. Firstly, centrifugation at 300 g for 10 min, followed by 2000 g for 10 min, and 10,000 g for 30 min. The supernatant was then filtered to an ultracentrifuge and centrifuged at 100,000 g for 70 min at 4°C twice to pellet the exosomes. Finally, exosomes were resuspended in PBS (usually 50 µL to 100 µL).
The shape and size of exosomes were observed by transmission electron microscopy (TEM) and size distribution of exosomes was detected by dynamic light scattering (DLS). The characterization of the isolated exosomes was confirmed by the expression of exosomal protein marker tumor susceptibility 101 (TSG101), CD63 and CD81.
Fluorescence in situ hybridization (FISH)
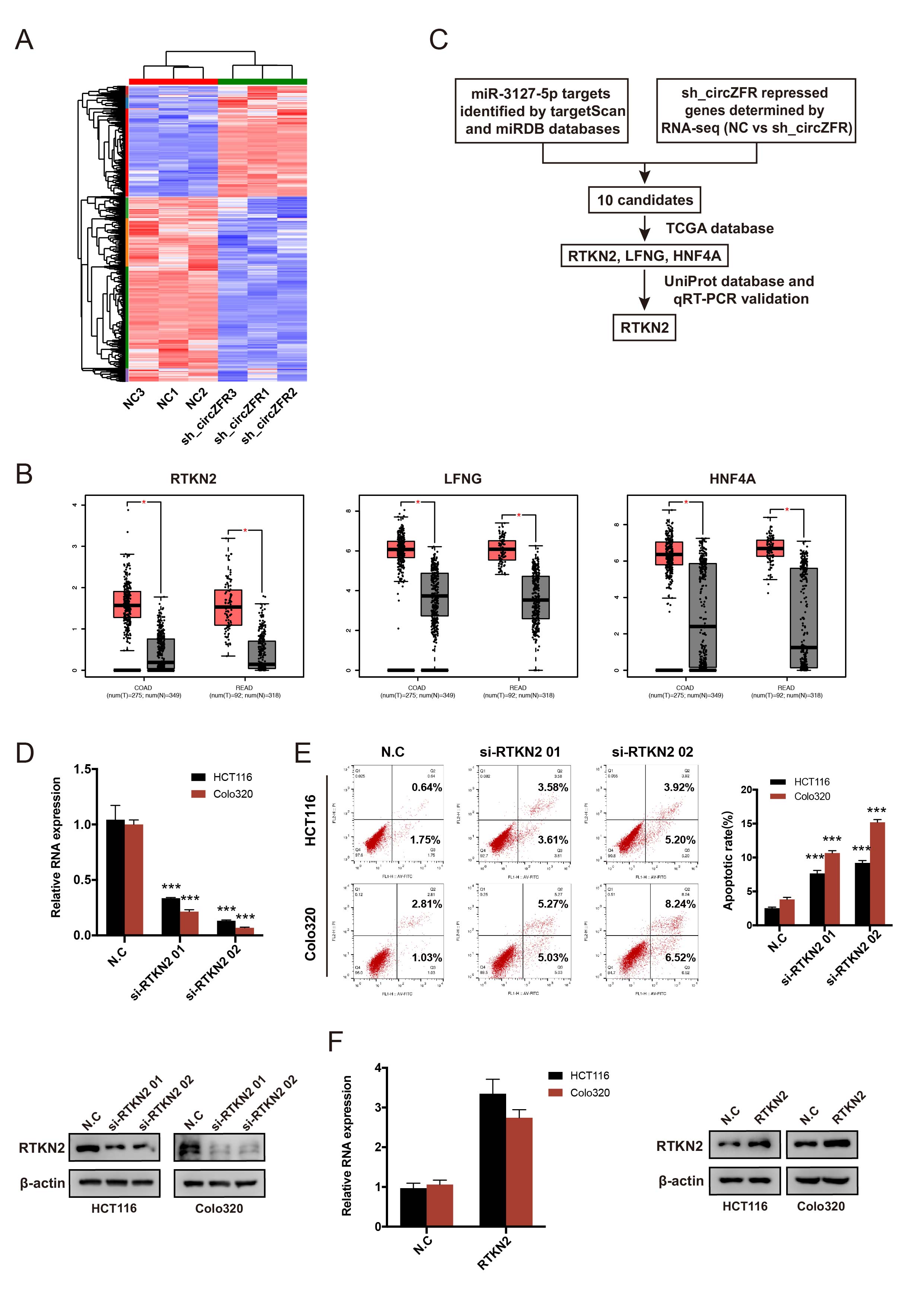
Digoxigenin (DIG)-labeled circZFR probes and Cy3-labled miR-3127-5p probes were designed and synthesized by RiboBio (Guangzhou, China). The FISH Kit (RiboBio, Guangzhou, China) was utilized for testing the expression and localization of circZFR and miR-3127-5p in CRC cells and the expression and localization of circZFR in TMA following the manufacturer’s guidelines. Images were captured under a fluorescence microscopy (Nikon, Tokyo, Japan) and the FISH signals were scored using Image-Pro Plus 6.0 (Media Cybernetics, Inc., Rockvile, MD, USA) software to obtain the average optical density of each photo.
SiRNA transfection and Lentivirus stable transduction
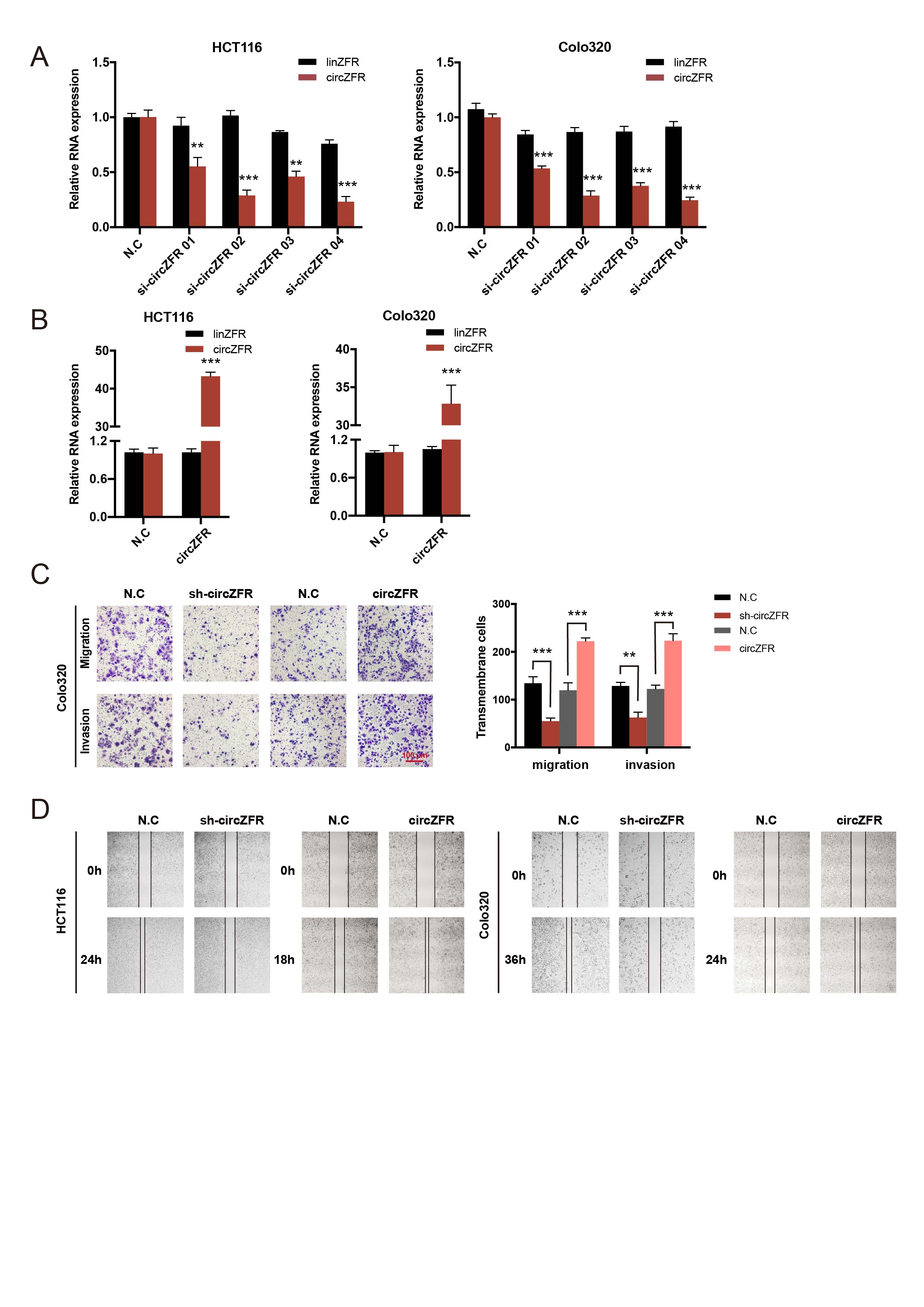
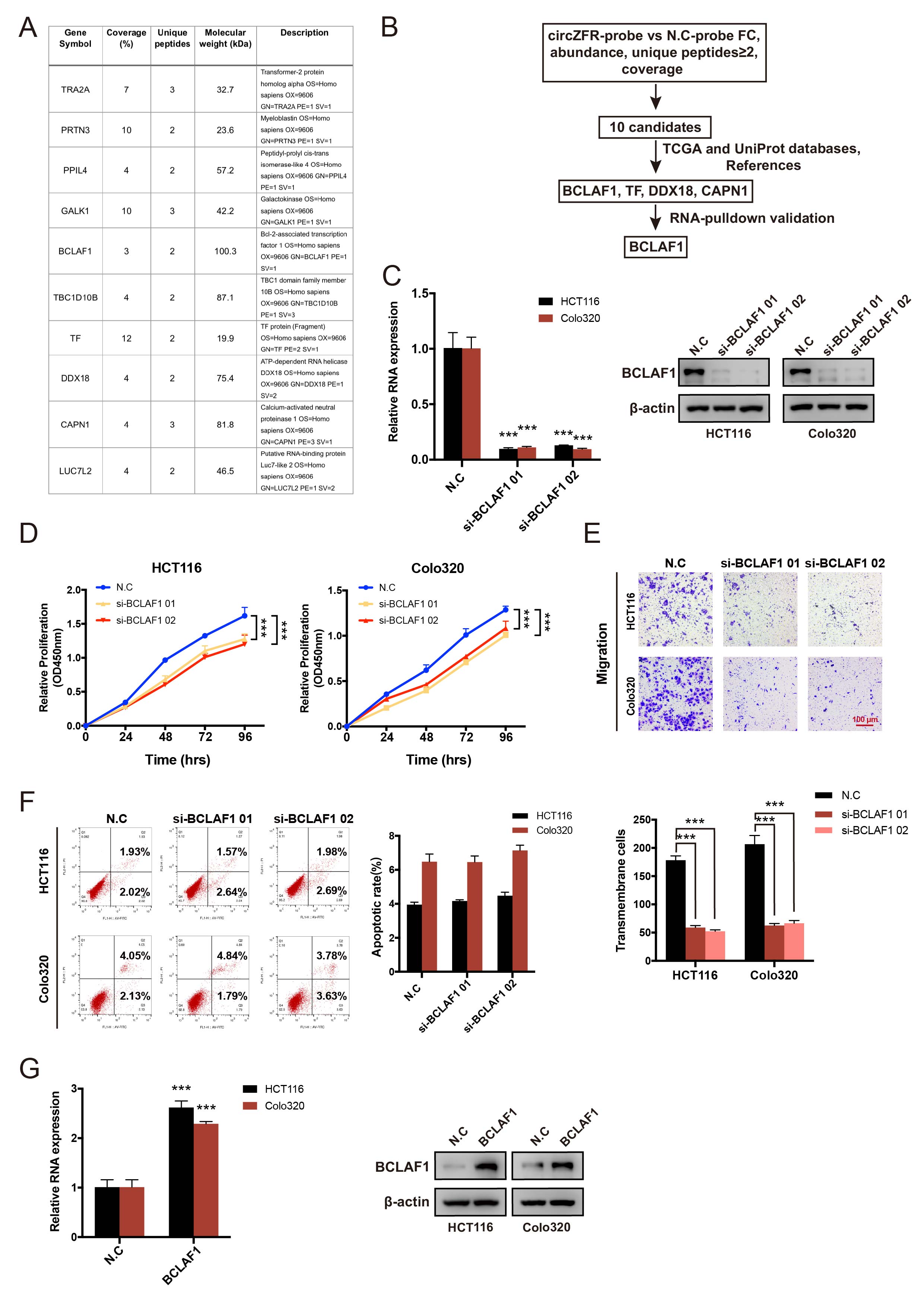
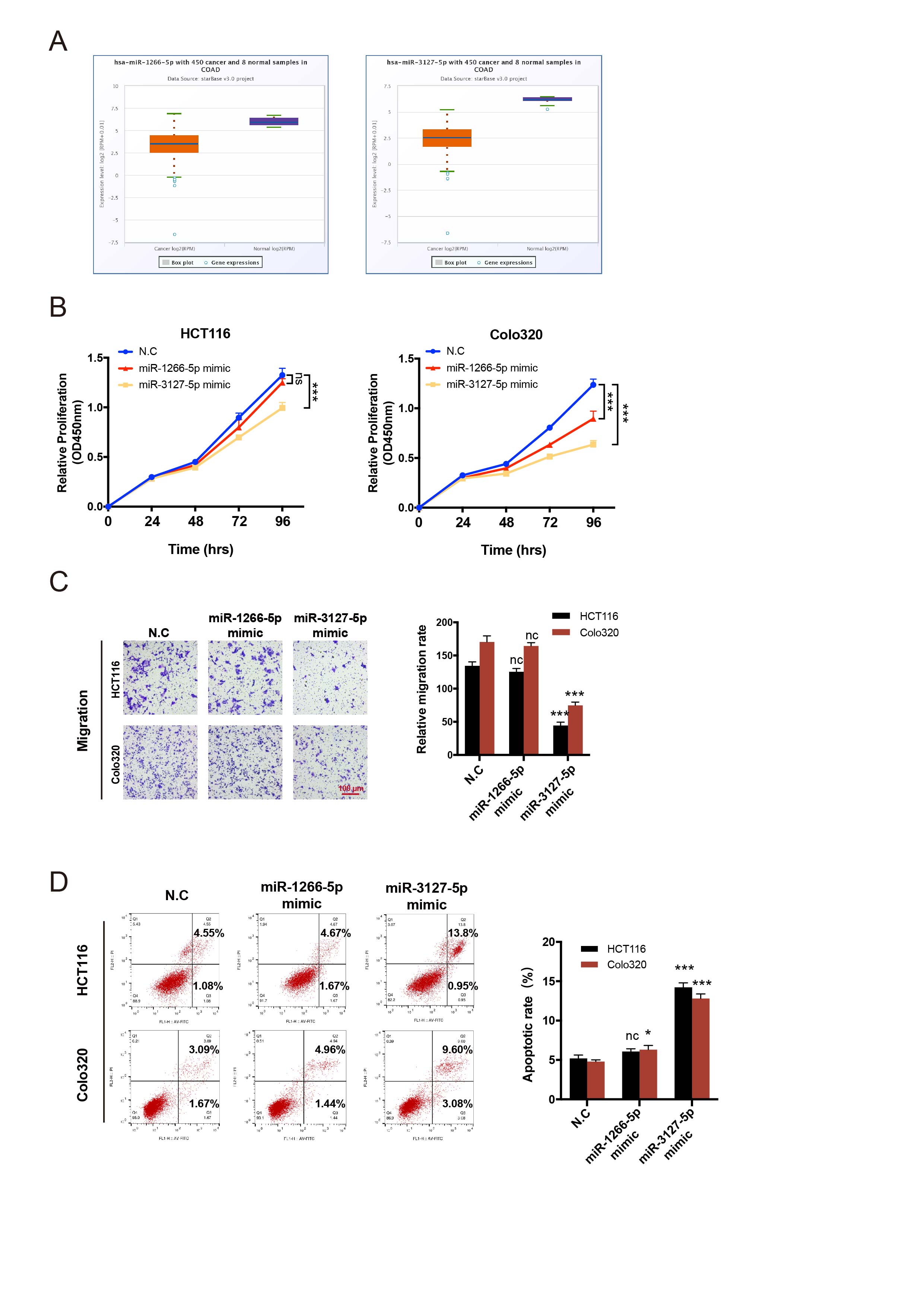
The human CRC cell lines HCT116 and Colo320, or 293T were seeded 40% confluent in 6-well or 24-well plates overnight. Small interfering RNAs (siRNAs) of circZFR (RiboBio, Guangzhou, China), miRNA mimics and inhibitors (GenePharma, Shanghai, China) were transfected by Lipofectamine RNAiMax (Invitrogen, CA, USA) following the manufacturer’s protocol. To construct small hairpin RNA (shRNA) or overexpression lentivirus plasmid, the sequence of si-circZFR or circZFR was cloned into the pHBLV-U6-MCS-CMV-ZsGreen-PGK-PURO vector or pHBLV-CMV-circ-EF1-ZsGreen-T2A-Puro vector (Hanbio Co. Ltd., Shanghai, China). The generation of lentivirus was achieved by co-transfection of the expression plasmid with the packaging plasmids psPAX2 and pMD2G using Lipofectamine 3000 (Invitrogen, CA, USA) according to the manufacturers’ instructions. After transfection, positive cells were propagated in 2 µg/mL puromycin (Gibco, Grand Island, NY, USA) for one week and harvested for subsequent experiments.
5-Ethynyl-2’-deoxyuridine (EdU) and Cell counting kit-8 (CCK-8) assays
The proliferation rate of CRC cells was detected by the Cell-Light EdU DNA Cell Proliferation Kit (RiboBio, Guangzhou, China) and the CCK-8 solution (Dojindo Lab. Kumamoto, Japan). For the EdU assay, cells were seeded 30%-50% confluent in 24-well plates and cultured for 24 h. EdU solution (50 µM) was added to each well of plate and incubated for 2 h. The cells were then fixed with 4% formaldehyde for 20 min and permeabilized with 0.5% Triton 100 for 10 min. In the following step, Apollo Dye solution to stain the EdU for 30 min and Hoechst 33342 to stain the nuclei. Images were obtained with a Nikon microscope (Tokyo, Japan) and the proportion of EdU-positive cells was counted by Image J.
For the CCK-8 assay, seven thousand cells were seeded in 96-well plates and 10 µL of CCK-8 solution was added to each well at the appointed time. After a 2-h incubation at 37°C in a 5% humidified CO2 atmosphere, the absorbance at 450 nm was measured with an automatic microplate reader (BioTek, Winooski, VT, USA). The EdU and CCK-8 experiments were performed in triplicate.
Soft agar colony formation assay
Primarily, semisolid agar medium (0.6% agarose/Phosphate buffer saline, PBS) was added onto the bottom layer of 6-well plates. Then, CRC cells were trypsinized and made into 25000 cells/mL suspension in culture medium. The mixture of CRC cells and semisolid agar medium (0.5% agarose/PBS) was added onto the bottom layer. After the gel was solidified, we added 1 mL culture medium on each well and changed the medium every 4 days. After at least 14 days of incubation, representative images of the cell colonies were obtained under microscope. The experiments were replicated at least three times.
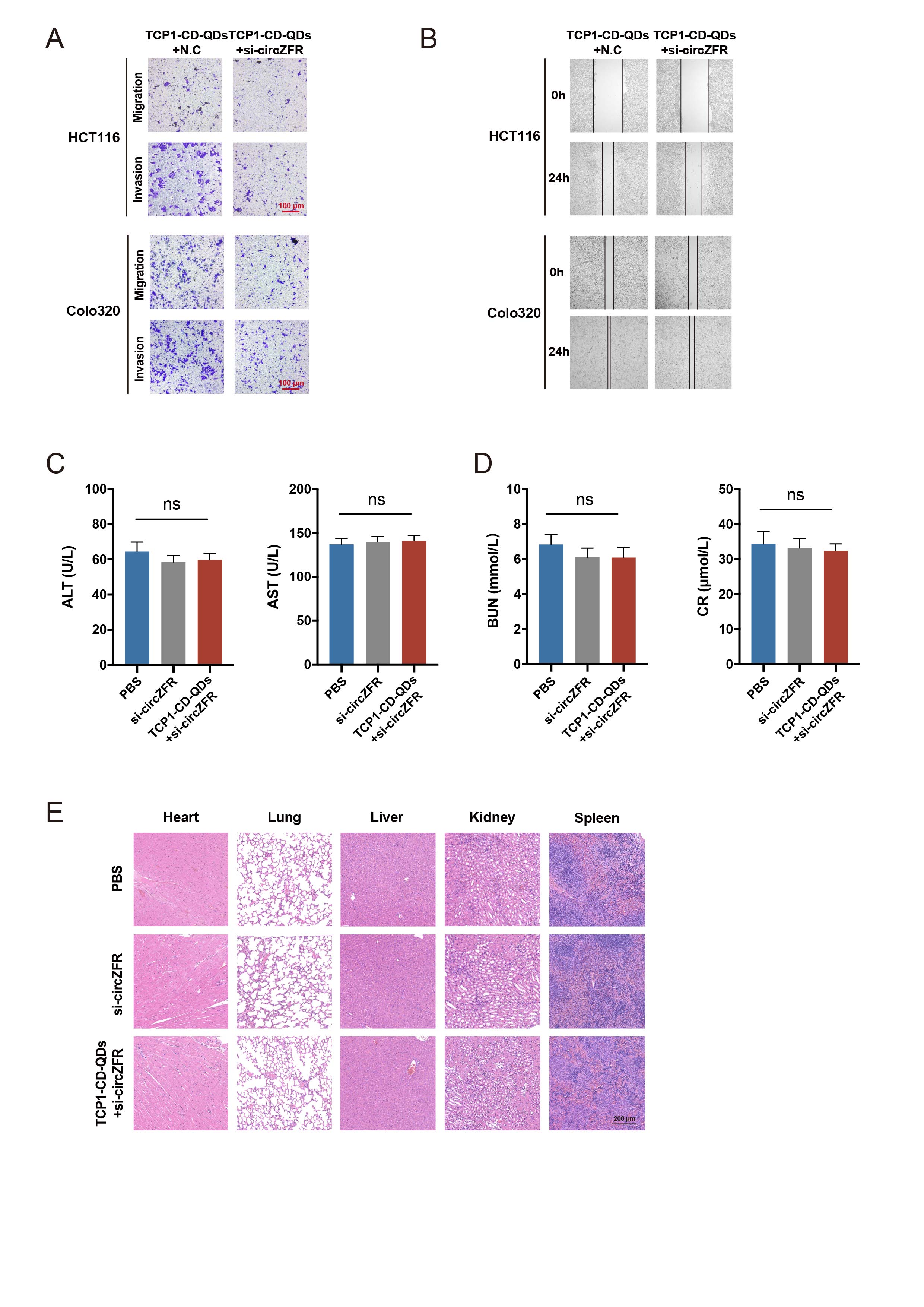
Transwell, wound-healing and Cytoskeleton assays
Transwell migration and invasion assays, and wound-healing assay were carried out as previously reported[22].
To visualize the actin cytoskeleton, chamber slides were primarily added in 12-well plates and then CRC cells were seeded in plates for 24 h. The PBS-rinsed cells were fixed with 4% formaldehyde for 20 min, permeabilized with 0.5% Triton 100 for 10 min and blocked with bovine serum albumin (BSA; Fude Bio, Hangzhou, China) for 1 h at room temperature. Alexa Fluor 594 Phalloidin (Thermo, Waltham, MA, USA) was used to mark Filament-actin (F-actin) to observe the cytoskeleton, and nuclei were stained with Hoechst 33342. Fluorescent imaging of cells was detected under an Olympus immunofluorescence microscope. All assays were independently performed in triplicate.
Apoptosis analysis
For detection of cell apoptosis, an Annexin V-FITC/propidium iodide (PI) Kit (BD Biosciences, SanDiego, USA) was used. CRC cells were isolated and washed with cold PBS for three times, then stained with Annexin V-FITC/PI at room temperature in the dark for at least 15 min. The apoptotic rate was analyzed using a flow cytometer (BD FACSCANTO Ⅱ, San Jose, USA) and FlowJo software. The experiments were repeated at least three times.
Western blotting and antibodies
Proteins in cells were extracted with RIPA lysis buffer (Fude Bio, Hangzhou, China). Western blotting experiments were performed according to details previously reported[22]. The primary antibodies used in this study are as follows: anti-E-cadherin (3195, Cell Signaling Technology, CST), anti-N-cadherin (sc-8424, Santa Cruz), anti-MMP9 (13667, CST), anti-Bcl-2 (4223, CST), anti-Bax (5023, CST), anti-cleaved caspase-9 (9509, CST), anti-cleaved caspase-3 (9661, CST), anti-β-actin (3700, CST), anti-TSG101 (ab133586, Abcam), anti-CD63 (ab134045, Abcam), anti-CD81 (ab109201, Abcam), anti-ESRP1 (21045-1-AP, proteintech), anti-BCLAF1 (ab181240, Abcam), anti-TF (17435-1-AP, proteintech), anti-DDX18 (ab128197, Abcam), anti-CAPN1 (2556, CST), anti-FLAG (14793, CST), anti-RTKN2 (Thermo, PA5-58392), anti-AGO-2 (MA5-23515, Thermo), anti-Ki67 (27309-1-AP, proteintech).
Biotin-labeled RNA pulldown and mass spectrometry (MS)
The 5’ biotin-labeled oligonucleotide probe targeting the junction site of circZFR was synthesized as follows: 5’- ACGACGUAAGGGCCAGCUUGGAAAAUCAG-3’. The biotin-labeled RNA probe was incubated with cell extracts from HCT116 cells stably transfected with N.C or circZFR overexpression vector. Then, the biotinylated circZFR-protein mixture was captured with Streptavidin Magnetic Beads and washed following the manufacturer’s protocol. The retrieved proteins were collected for Western blotting detection or silver staining (Beyotime, Shanghai, China) and mass spectrometry (MS) analysis.
RNA-protein immunoprecipitation (RIP) and IP
The RIP experiments were performed by using a Magna RNA-binding Protein Immunoprecipitation Kit (Millipore, Bedford, MA, USA) according to the manufacturer’s protocol. In brief, HCT116 extracts were incubated with protein A/G beads conjugated to an antibody against ESRP1 or BCLAF1 or AGO-2 and controlled by normal rabbit IgG (A01008, GenScript).
N.C/circZFR overexpressing HCT116 cells were transfected with FLAG-tagged BCLAF1 overexpression plasmid transiently by Lipofectamine 3000 (Invitrogen, CA, USA). After treatment of MG132 for 12 h, cells were harvested and lysed, then incubated with anti-FLAG M2 Magnetic beads (Sigma, MO, USA). After rotating incubation overnight at 4°C, the bead-bound proteins were released and detected by Western blotting with an anti-ubiquitin antibody (3936, CST) according to the manufacturer’s protocol.
Dual luciferase reporter assay
The binding between circZFR and miRs was detected by the Dual luciferase reporter Kit (Beyotime, Jiangsu, China). The wild type (WT) or mutant type (MUT) circZFR sequence was cloned into the PSI-Check2 vector (Hanbio Co. Ltd., Shanghai, China), which contains the firefly luciferase gene and Renilla luciferase gene. For the RTKN2 and miR-3127-5p experiments, WT or MUT RTKN2 3’UTR sequence was cloned to the PSI-Check2 vector. 293T or CRC cells were seeded 50% confluent in 24-well plates overnight and then cotransfected with circZFR-WT/MUT luciferase vector or RTKN2-WT/NUT luciferase vector and PSI-Check/miR mimics. After 24 h, the luciferase activity was measured according to the manufacture’s protocol.
Animal studies
Firstly, luciferase-expressing HCT116 cells were stably transfected with N.C or sh-circZFR vectors to construct the N.C or sh-circZFR group. For the subcutaneous xenograft tumor model, 12 BALB/c nude mice (male, 4 weeks old) were randomly divided into 2 groups, and each was implanted subcutaneously in the flank site with 4×106 cells suspended in sterile PBS. Three weeks later, tumors were imaged using a Spectrum in vivo imaging system (IVIS) (PerkinElmer, USA). For the pulmonary metastasis model, 12 mice (male, 5 weeks old) were randomly divided into 2 groups, and each was injected into the tail vein with 2×106 cells suspended in sterile PBS. Tumors were imaged after four weeks. For the hepatic metastasis model, 12 mice (male, 6 weeks old) were randomly divided into 2 groups, 4×106 cells suspended in PBS were injected into the inferior of spleen. Tumors were imaged after five weeks. The in vivo imaging of mice was performed according to the previously described protocols[22]. After imaging, the mice were sacrificed and the tumors and organs including lung and liver were harvested and used for H&E and IHC staining according to the manufacturer’s protocol. All the animal studies were approved by the Zhejiang University Animal Care and Use Committee and performed following the national guidelines and regulations.
To construct the PDX models, fresh CRC tissues were cut into 2 mm3 pieces and washed twice with PBS and suspended in RPML 1640 medium containing penicillin and streptomycin. Then, the tumor specimens were placed in the flank of 15 nude mice. When the tumor volume reached 100 mm3 (volume = 0.5 × width2 × length), mice were randomly divided into 3 groups and intravenously injected with PBS, si-circZFR conjugated with cholesterol (Chol) or TCP1-CD-QDs loaded with si-circZFR every three days. The dosage of siRNA in the Chol-conjugated si-circZFR group and nanocarrier group were based on the loading capacity of TCP1-CD-QDs (2 mg/kg). After three weeks, the mice were sacrificed and tumors were harvested for further H&E and IHC staining. All patients provided consent to allow their tissues to be stored and used for research. This study was approved and monitored by the Ethics Committee of the Sir Run Run Shaw Hospital, Zhejiang University.
Statistical analysis
Data are presented as the means ± standard deviations (SDs). Paired or unpaired two-tailed Student’s t-tests and one-way ANOVA followed by Bonferroni tests were applied for the group comparison. Chi-square calculations or one-way ANOVA was used to assess the relationship between circZFR expression and clinicopathological characteristics of CRC patients from TMA. The receiver operating characteristic (ROC) curves were applied to assess the diagnostic capacity of circZFR in tissues and serum from CRC patients with different clinical characteristics. The Kaplan-Meier analysis was used to evaluate the effect of circZFR on the overall survival (OS) of CRC patients from TAM. Univariate Cox regression analysis was utilized to estimate hazard ratios (HRs) and corresponding 95% confidence intervals (CIs). The correlations between circZFR and ESRP1 or RTKN2 in CRC tissues was determined by Pearson’s correlation analysis. Statistical analysis was performed by SPSS software and GraphPad Prism 8 software, and significance was set at *p < 0.05, **p < 0.01 and ***p < 0.001.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}