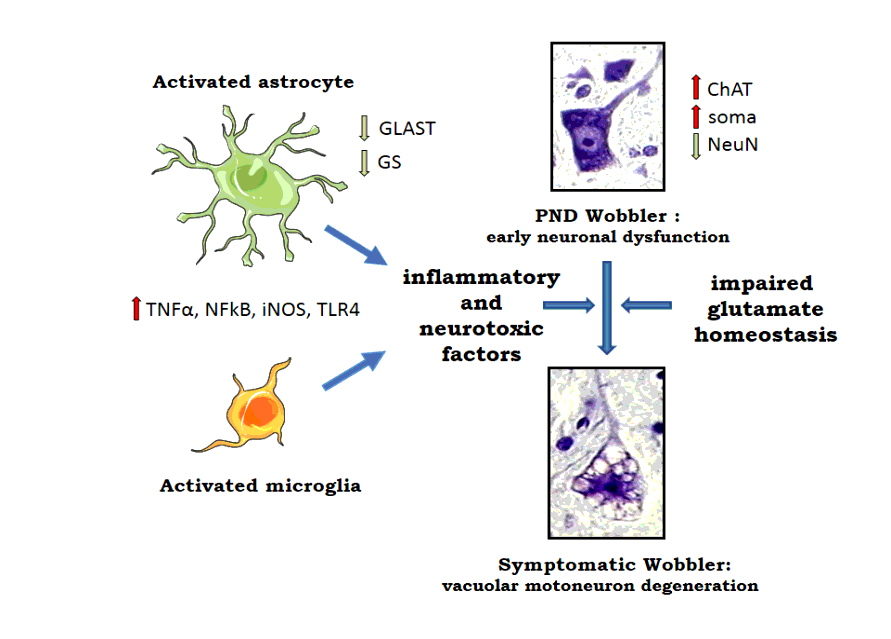
Experimental animals
Newborn mice from the parental NFR/wr strain were genotyped from a tail biopsy, using published procedures (Meyer et al. 2015; Schmitt-John 2015). On PND 6 genetically identified Wobbler mice (wr/wr) and wild type control mice (NFR/NFR) were used for the different experiments. Because neither disease onset nor the disease progression differ between male and female Wobbler mice (Meyer et al. 2015) mice of both sexes were used in similar numbers in all experiments. Wobblers comprised ~20% of each litter, which made necessary to mate large number of breeding pairs. For immunocytochemistry pups were anesthetized with a mixture of ketamine (75 mg/kg, i.p.) and xylazine (6 mg/kg, i.p) and perfused intracardially with 2.5 ml of 0.9% NaCl, followed by 4% paraformaldehyde (PFA) in 0.1 M sodium phosphate buffer pH 7.2. Afterwards spinal cords were dissected, postfixed for 2.5 h at 4oC in the same fixative and finally embedded in paraffin. Five µm sections of cervical spinal cords were cut in a microtome and placed on microscope slides. For immunofluorescence, cervical spinal cords from heart-perfused mice were embedded in Tissue-Tek (OCT compound, Miles Inc., USA), and 16-30 µm sections cut in the horizontal plane in a cryostat maintained at -20 oC. For mRNA analysis by qPCR, mice were killed by decapitation and spinal cords stored at -80 o C until used for RNA extraction. About 5-6 samples were studied per mice in all measurements.At this early age period, anatomical or behavioral differences were not apparent between control and Wobbler mice. To prevent bias in the results of immunocytochemistry, immunofluorescence and PCR analysis, researchers were blinded to experimental protocols.
Immunocytochemistry for labeling astrocytes and microglia
For astrocyte staining in spinal cord sections, we used a rabbit polyclonal glial fibrillary acidic protein (GFAP) antibody (1:250 dilution, # G-9269 from SIGMA-Aldrich, MO, USA), followed by a goat anti-rabbit IgG conjugated to Alexa Red 555 (1:1000 dilution) (Invitrogen, Molecular Probes, Eugene, OR, USA). The final step included drying of sections at room temperature and cover slipped with Fluoromount-G (Southern Biotech, Birmingham, AL, USA). GFAP + cells were counted in the ventral horn gray matter. Images produced by confocal microscopy were analyzed using Image J (Image Processing and Analysis in Java, NIH, MD, USA) at 200X from at least 6 sections per mice . The number of GFAP+ astrocytes was quantified by this program and results were averaged per mice. Data was expressed as the mean number of GFAP+ cells ± SEM per unit area (mm2). The quantity of Iba1+ microglia was determined by immunofluorescence staining (Meyer et al. 2015). Spinal cord sections from PND 6 Wobbler and control pups embedded in Tissue-Tek were stained for resting and reactive microglia using a rabbit anti-Iba1 antibody (1:1000, cat. #019-19741, Wako, Japan). The secondary antibody was a goat anti-rabbit IgG conjugated to Alexa Red 555. Sections were cover-slipped with Fluoromont-G. Iba1+ immunofluorescent microglial cells were counted in the gray matter of the ventral horn of the spinal cord (Meyer et al. 2015) using methods described above for GFAP+ astrocytes. The number of Iba1+ microglia was expressed per unit area (mm 2). Cells were counted in 5-6 sections from each animal. In addition to cell density, we analyzed the morphological immunophenotype of Iba1+ cells. For this purpose, cells were classified as ramified, showing small body with long processes, or ameboid with round body and scarce short processes.
Immunocytochemistry for labeling Glutamine synthase (GS) and GLAST
For GS, 5-µm-thick spinal cord sections were treated with mouse IgG blocking reagent (Vector M.O.M. Immunodetection Kit, Vector Labs, Burlingame, CA, USA), washed and incubated overnight with a 1/200 dilution of a purified monoclonal mouse anti-GS (#610517, BD Biosciences). As second antibody we used a 1/1000 dilution of a goat antimouse IgG conjugated to Alexa Green 488 (Invitrogen). Sections were cover-slipped with Fluoromount-G and the number of GS+ cells counted using Image J program. For GLAST staining sections were first blocked with 1% H2O2, then with 10% goat serum in PBS and exposed to a primary goat EAAT1 polyclonal antibody (sc-7757, Santa Cruz Biotech, TX, USA). The second antibody was a rabbit anti-goat from Sigma (cat.7014). Sections were then processed according to the ABC kit instructions (Vector). Determination of GLAST immunoreactive was carried out by computerized image analysis, as detailed in Meyer et al (2018).
Double immunofluorescence staining for GFAP and GS
To study if GS colocalized with GFAP in control and Wobbler astrocytes, 5 µm thick spinal cord sections were treated with mouse IgG blocking reagent (Vector M.O.M. Immunodetection Kit), washed and incubated overnight with a 1/200 dilution of a purified monoclonal mouse anti-GS (#610517, BD Biosciences, Franklin Lakes, NJ , USA.). As second antibody, we used a 1/1000 dilution of a goat antimouse IgG conjugated to Alexa Green 488 (Invitrogen). Afterwards, staining of astrocytes was performed with a rabbit GFAP antibody (1/250 dilution; same antibody shown above). Sections were then incubated with a goat anti-rabbit IgG conjugated to Alexa Red 555 (Invitrogen). The final step included drying of sections at room temperature and cover slipped with Fluoromount-G. This antibody combination produced green and red fluorescent labeling of each antigen, respectively. Dual-labeled immunofluorescence microscopy was analyzed with an Olympus IX83 inverted microscope equipped with a disk-spinning unit (Olympus Corporation, Japan). Digital images were acquired using Cell Sens Dimensions software from Olympus and photographed with a Hamamatsu Orca Flash 4.0 monochromatic camera (Hamamatsu Photonics K.K, Japan).
Gene expression in PND 6 control and Wobbler mice
Analysis of mRNA expression by qPCR was performed following published procedures (Garay et al. 2012; Meyer et al. 2018). Total RNA from the spinal cord of control and Wobbler pups was extracted with Trizol (Life Technologies-Invitrogen, CA, USA), and residual DNA degraded by deoxyribonuclease 1 (DNase1, EC 3.1.21.1) (Promega, Madison, WI, USA) treatment. A M1705 MMLV reverse transcriptase (EC 2.2.2.49; Promega) was used for PCR amplification of DNA templates in the presence of random hexamer primers. Sequences of forward and reverse primers for the microglial marker CD11b and proinflammatory factors TNFα, iNOS, TLR4 and p65NFkB were those previously published (Garay et al. 2012; Meyer et al. 2018). Cyclophilin was used as the house keeping gene. Gene expression profiles were assessed by a real Time Step-one Plus sequence Detection System (Applied Biosystems, Foster City, CA, USA) and analyzed by the 2 −ΔΔCT method (Livak and Schmittgen 2001). Results were expressed as fold induction over control.
Immunocytochemistry for choline acetyltransferase (ChAT).
For ChAT immunostaining (Meyer et al. 2010), 16 μm cryostat sections of the spinal cord were postfixed in 4% paraformaldehyde and kept at −80°C until used. After defrosting, sections received 1% H2O2 in methanol to block endogenous peroxidases and incubated at 4°C overnight with a 1/500 dilution of a ChAT goat polyclonal antibody (AB 144P, Chemicon, CA, USA). Sections were then incubated for 60 min with a biotinylated rabbit anti-goat secondary antibody (1/200 dilution) and then with a 1/100 dilution of the ABC complex (ABC kit, Vector Labs, CA, USA). The peroxidase activity was revealed using diaminobenzidine (DAB, 0.25 mg/ml) in the presence of 0.01% H2O2 in the dark. The number of ChAT+ cells was quantified in 10 sections per animal in the ventral horn using a computerized image analysis system (Bioscan Optimas 6.02). Digital images (digital camera Panasonic GP-KR222 connected to an Olympus BH2 microscope of tissue sections were displayed on the video screen under identical lighting conditions and grey-scale threshold.
Cresyl violet staining for light microscopy
Following laminectomy, cervical spinal cords were removed and small blocks of tissue were obtained by cutting transverse sections of 2–3 mm maximum length. Blocks were immersed for 2.5 h in 4% PFA, postfixed in graded series of ethanols and embedded in paraffin. Paraffin sections (5 µm) were hydrated and stained with cresyl violet (0.5% aqueous solution) and then dehydrated in graded ethanols and xylene, and mounted with Permount.
Immunofluorescence staining for NeuN
The phenotype of neurons of the ventral spinal cord was investigated using immunostaining for NeuN. 30-µm cryostat sections were blocked with 3% donkey serum in PBS 0.5% Triton X-100 for 10 min at 37 °C, then incubated overnight with the monoclonal NeuN antibody (anti-Neuronal Nuclei MAB 2377, Chemicon-Millipore, Billerica, CA, USA) at a 1/200 dilution. Slices were rinsed three times in PBS 0.1% Triton X-100 for 5 min before application of the second antibody donkey anti-mouse IgG conjugated to Alexa Fluor 488 (dilution 1/500). Incubation with the second antibody was followed by three rinses in PBS. Sections were mounted with Fluoromount G and kept in the dark at 4 °C until analysis by confocal microscopy. Cells were examined under an Olympus IX83 inverted microscope equipped with a disk-spinning unit (Olympus Corporation, Japan). Digital images were acquired using Cell Sens Dimensions software from Olympus and photographed with a Hamamatsu Orca Flash 4.0 monochromatic camera (Hamamatsu Photonics K.K, Japan). Quantification was performed using the Image J system, as explained above.
Double immunofluorescence analysis of ChAT/NeuN+ cells by confocal microscopy
To localize ChAT with NeuN immunoreactive motoneurons, 30 μm cryostat sections were heated for 10 min at 120 °C to retrieve antigens in 10 mM sodium citrate buffer pH 6.0. Afterwards, sections were blocked with 3% donkey serum in PBS for 10 min at 37 °C and incubated overnight at 4 °C with 1/100 dilution of the ChAT goat polyclonal antibody (AB 144P) and NeuN antibody (anti-Neuronal Nuclei MAB 2377, Chemicon-Millipore, Billerica, CA, USA) at a 1/200 dilution in 2% donkey serum, and 0.5% Triton-X100 in PBS. Slices were rinsed three times in PBS 0.1% Triton X-100 for 5 min before application of the second antibody donkey anti-goat IgG conjugated to Alexa Fluor 555 (dilution1/500) and donkey anti-mouse IgG conjugated to Alexa Fluor 488 (dilution 1/500). After several washes in PBS, sections were mounted with Fluoromount-G (0100-01), and examined under an Olympus IX83 inverted microscope equipped with a disk-spinning unit (Olympus Corporation, Japan). Confocal images were acquired, photograph and analyzed as described above for NeuN cells. We determined double-labeled ChAT+/NeuN+ neurons in the ventral horn and results expressed as the percentage of ChAT+/NeuN+ neurons per unit area (mm2).
Statistical analysis
Results were expressed as mean ± S.E.M. Data were analyzed by two-tailed Student`s “t” test since only two groups (control and Wobbler) were compared. Statistical analyses were performed with Prism 6 GraphPad software (San Diego, CA, USA) and significance was shown by the following asterisks: *p < 0.05, **p < 0.01, and ***p < 0.001.
{kind=link}