Design and Setting
We conducted this non-inferiority randomized trial at Centre hospitalier universitaire de Sherbrooke (CHUS, performing over 200 RCB per year) and Centre Hospitalier de l’Université Laval (CHUL, where this method was recently implemented), Quebec, Canada between September 2016 and May 2017. The trial was approved by the research ethics board of Centre intégré universitaire de santé et service sociaux de l’Estrie (MP-31-2017-1298), which provided a provincial-wide approval. Clinical Trial Registration: ClinicalTrials.gov (NCT02913625). All study participants provided written informed consent before randomization. A detailed study protocol was previously published (10, 13). This study fully adhere to CONSORT guidelines (14).
Study participants
All patients scheduled for elective or urgent surgery (mainly open reduction and internal fixation of arm and forearm, fasciectomy, epicondylitis, elbow and wrist arthroscopy and elbow arthroplasty) were eligible for the study if they were ≥18 years old; ASA (American Society of Anesthesiologists) class I-III; able to provide a valid written consent and weighed > 50 kg, regardless of body mass index (BMI). Patients with previous surgery or gross anatomical deformities of the clavicle, systemic or local infection at the needle entry point, coagulopathy, severe pulmonary disease, local anesthetic (LA) allergy, known neuropathy affecting the operated limb, pregnancy and for whom surgeon requested an indwelling catheter for postoperative analgesia were excluded.
Intervention
In the intervention group, an RCB was performed by inserting the needle at the supraclavicular fossa and aiming the posterior wall of the axillary artery, strictly in-plane with the US probe resting in the delto-pectoral groove (10). For the control group, the ICB was performed by placing the US probe in the infraclavicular fossa, medially to the coracoid process and by directing the needle towards the posterior wall of the axillary artery. Local anesthetic bolus (20 ml of 0.5% ropivacaine and 20 ml of 1.5% mepivacaine) was deposited under the axillary artery for both approaches. As defined a priori, ICB and RCB feasibility needed to be confirmed by a pre-scan before the randomization envelope was opened, to avoid recruitment of patients with gross anatomical deformation. Objective evaluation of motor and sensory blockade was required for all patients.
Randomization and Blinding Process
Clinical and epidemiological research unit at the CHUS generated the random allocation sequence. Participants were randomly assigned to control or experimental group with a 1:1 allocation ratio and stratified by sites using permuted blocks of random sizes. To ensure concealment, block sizes will not be disclosed and sequentially numbered, opaque and sealed envelopes were used. Only the research assistant had access to the envelopes. A research assistant, resident or anesthesiology department staff member was responsible for assessing participant eligibility and recruitment. All anesthetic interventions were performed by an anesthesiology resident (minimally in its 2nd year of training) or an anesthesiologist of the recruiting centers. Before the study, all operators, including experienced attending anesthesiologists, had a minimum experience of three successful retroclavicular blocks, as well as a minimum overall experience of 20 regional blocks. Only the outcome assessor for the motor block and sensory block could be blinded since it was technically impossible to blind the person performing the block and the patient. To reinforce blinding of the motor and sensory block assessor, chlorhexidine and bandages were applied over the two theoretical needle entry points. Video assessors of needle visibility could not be blinded because needle position on the screen was evident, but the adjudication was made independently to minimize this bias.
Outcomes Assessment
A research assistant recorded the different times needed using a standard chronometer. Imaging time was the interval between initial contact of US probe with the skin and acquisition of a satisfactory axillary artery image. Needling time was the interval between the skin wheal and block needle withdrawal. Total anesthesia time was the sum of performance time (imaging plus needling) and the time required to achieve a sensory loss score of 9/10 (further described). The primary outcome, performance time, was measured in minutes (min) and corresponded to the sum of imaging time and needling time.
Detailed methodology of the following outcomes can be found in our previously published protocol (13). Briefly, depth of sensitive and motor blocks was evaluated every 10 min after complete LA injection. Sensory loss was assessed in the territory of the radial, median, ulnar, musculocutaneous, and medial cutaneous nerve of the forearm using a 3-point score. As previously described (10, 15), the sum of these five scores represents the final sensory loss score (minimum score of 9/10 required). Motor function was tested for the radial, median, ulnar, and musculocutaneous nerves. The sum of these four scores represents the motor block final score (10, 15). An independent, blinded, research assistant completed the sensory and the motor assessment and noted the final scores up to 30 min after block was performed. Blocks were considered successful only if surgery was completed without additional LA infiltration or GA. Blocks were recorded on video and two blinded independent anesthesiologists evaluated needle visibility using a 5-point Likert scale and ImageJ software (version 1.50i, developed by the National Institutes of Health [NIH]). This evaluation was done twice and needle angle on the horizontal axis was measured and noted before LA injection.
Immediately after complete LA bolus injection, an independent and blind outcome assessor evaluated procedural pain using a 10 cm visual analogue scale. Following our detailed protocol (13), number of needle passes (defined as positive integer unit each time the block needle needs to be realigned on the skin) as well as early and 48h complications (such as paresthesia, vascular puncture, Horner syndrome, dyspnea, etc.) were also documented. We considered the use of neurostimulator as positive when the purpose of its use was not only for safety sentinel stimulation (<0.3 mA). Details of complications and follow-up assessments are presented in our protocol (13). Briefly, early complications were assessed on the day of surgery, all patients were further contacted 48h after surgery to query for any delayed complications (dyspnea, paresthesia, weaknesses, pain, at the puncture site or hematoma). If a complication was suspected, patients were immediately referred for medical assessment.
Statistical Methods
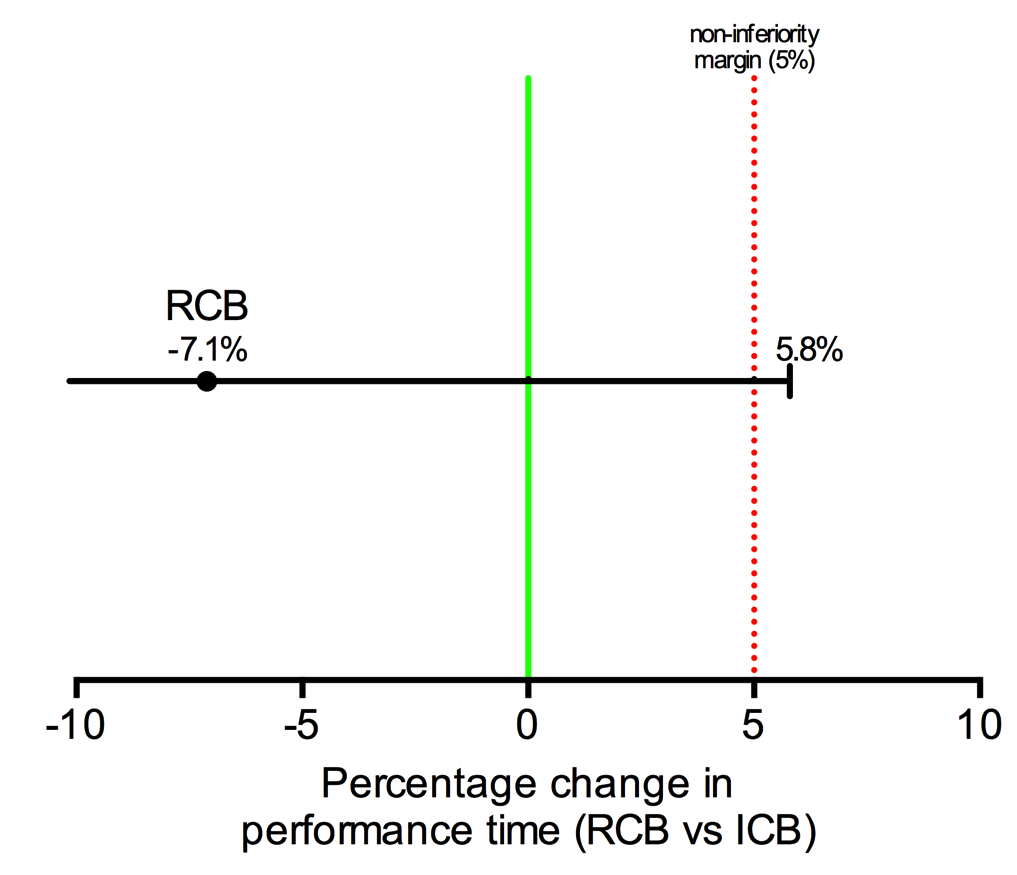
The statistics software SPSS (version 24.0.0.0), SAS (version 9.4), R (version 3.5.1) and Graph Pad Prism (version 6.0h) were used. Primary outcome was analyzed with the non-inferiority test of the averages. Data was not normally distributed and required a logarithmic transformation to perform pre-defined parametric tests. Primary outcome was interpreted based on the logarithmic transformation. Secondary outcomes were analyzed using superiority analysis. For continuous data or ordinal data with >8 categories, data were compiled as averages and standard deviation. Student t test was used for parametric data and the Mann-Whitney test was used for non-parametric data. Chi square (data n>5) or Fisher exact (data n <5) test were used for dichotomous data. Finally, for ordinal parametric data the chi square test was used and the Mann-Whitney test was used for ordinal non-parametric data. Subgroup analysis were performed following the same criteria as the main group and was conducted to evaluate if BMI higher than our study population average was influencing the outcomes. For all analysis, the intention-to-treat principle was used for analysis of missing data. When required, results are reported as mean (SD). No interim analysis was performed.
Sample Size
Sample size calculation for this study has been presented in detail previously (13). Briefly, it was calculated on the basis of a recent study (16) where the performance time for coracoid ICB approach was 5.6 min, with 45 s of visualization time and using our feasibility study recently conducted (10) where the needling time for the RCB approach was 3 min 42 sec. As we explained in our published protocol (13) and based on our clinical judgement and statistical convention, we deemed that a time superiority of 5% would be significant and therefore have set the non-inferiority margin at 5%. An initial sample size of 49 patients per group was required to provide a statistical power of 0.9 (0.05 one-sided type 1 error), but to account for dropouts and inadequate procedures, the final sample size was set to 55 patients per group.
{kind=link}