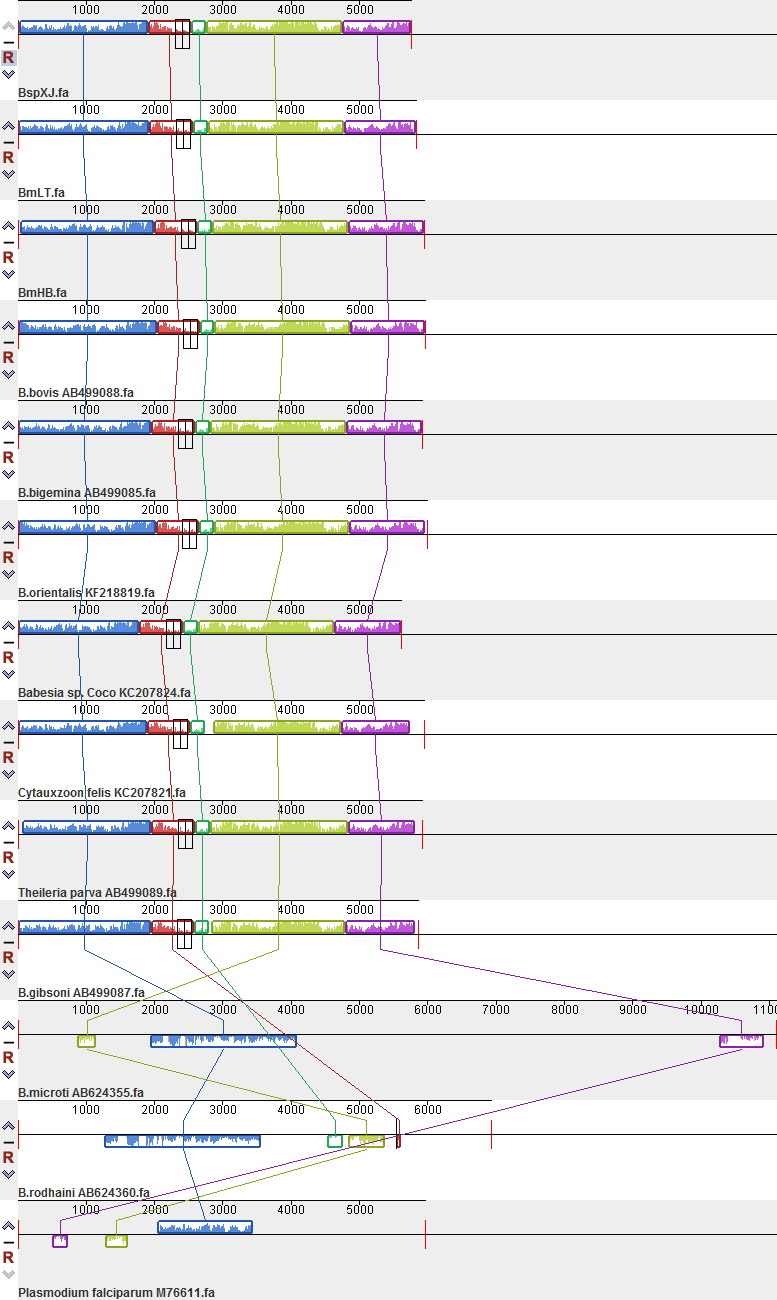
In the present study, we assembled and annotated the mitochondrial genomes of six ovine Babesia isolates and performed a mitochondrial genomic analysis with published mitochondrial genomes of apicomplexan parasites. The mitochondrial genomes of the six Babesia isolates infective to small ruminants are highly similar to those of most Babesia spp. with respect to genome size, high A + T content, genome form, and gene content. However, they are smaller than those of B. microti, B. rodhaini and T. equi and larger than that of T. gondii [28, 29, 44]. Consistent with the results from other apicomplexan parasites, tRNA genes were not found in the mitochondrial genomes. We speculate that they may be directly encoded by the nuclear genome. The order and transcriptional direction of three protein-encoding genes are the same in B. bovis, B. bigemina and B. gibsoni [28] but different from those in B. microti, T. equi, T. orientalis and P. falciparum [28, 29].
In the reported mitochondrial genomes of piroplasms, B. microti and B. rodhaini have a dual flip-flop inversion system, ranging from 184 to 1082 bp in length [29]. However, one pair of TIRs was found in the mitochondrial genomes of other Babesia spp. and Theileria spp., ranging from 25 to 1563 bp in length [26–28, 30, 32]. These findings indicated that the number and size of TIRs are one of the main causes of different mitochondrial genome sizes. We also found that different numbers of LSUs are responsible for the size of the mitochondrial genomes. TIRs are considered to play a crucial role in the replication and stabilization of linear mitochondrial genomes [45]. In the published mitochondrial genomes of apicomplexan parasites, the sequences of the coding genes and LSU are basically the same. Therefore, we speculated that differences in the lengths and sequences of TIRs may be responsible for divergence in the host-specific and in vitro culture characteristics of protozoans.
The difference between the Illumina and Sanger sequencing data is mainly caused by nucleotide substitutions, deletions and insertions, which result from the use of different sequencing techniques assembly and annotation software. The settings of the parameters are different, which is one of the reasons for the difference between the two datasets. The results of the Illumina sequencing method are more prone to errors than those of the Sanger method. The Sanger approach is more accurate and thus more appropriate for further studies of small genome sequencing.
Mitochondria are essential organelles and play an important role in energy metabolism, growth and development of apicomplexan protozoa [46]. Cytochrome bc1 is an integral membrane protein complex that is vital to cellular respiration. The highly conserved binding site of inhibitors in cytochrome bc1 is the molecular basis of the drug effect on yeast, fungi and parasites [43]. The drug binding residues in the cytb sequences of six ovine Babesia isolates are completely consistent. In the conserved PEWY region, Phe278 (yeast number of cytb) is present in most organisms, whereas T. gondii and B. taurus have Tyr and Ala, respectively, at this position. Studies have reported that the L275F mutation in yeast has no effect on enzyme activity, but the IC50 increased ten-fold [22]. Compared with the wild-type yeast, the Y279S mutant had a 40-fold increased IC50 (B1.7 mM) for atovaquone [22], and Y268S in the P. falciparum numbering system resulted in a 3000-fold loss of atovaquone sensitivity. In addition, the mutations M133I, L271V and Y268N of P. berghei confer resistance to atovaquone [43]. Therefore, we conclude that mutations in cytb are largely responsible for the efficacy of drugs (the target protein is the bc1 complex) in apicomplexan parasites.
Currently, atovaquone and ELQs have been reported for the treatment of human babesiosis and malaria by modifying the drug target through disruption of the cytochrome bc1 complex [19, 21, 22, 41, 43]. In 2019, a B. motasi-like parasite was detected in human blood in Korea [47], which suggests that B. motasi may be potentially zoonotic. Therefore, we should investigate the infection of B. motasi in humans in China and evaluate the zoonotic potential of B. motasi and the effect of inhibitors binding to the cytochrome bc1 complex. Our data showed that atovaquone, stigmatellin, myxothiazol, endochin-like quinolone (ELQ), antimycin A and NQNO drugs can be used in the treatment of babesiosis in the future. The molecular mechanism of the resistance of these drugs is the mutation of cytb, which suggests that a combined drug strategy is possible to avoid drug resistance during treatment of babesiosis.
In this study, the taxonomical relationships of Babesia isolates infective to sheep and goats are consistent with the reported phylogenetic analyses based on cytb, cox1, cox3, nuclear small subunit (SSU) and internal transcribed spacer (ITS) [5, 6, 8, 32, 48]. The ovine Babesia isolates are divided into two species: Babesia sp. and B. motasi. Babesia motasi further fell into two small clades, named BmLT/TZ and BmNX/HB. With the exception of B. conradae, piroplasm infective to the same host fell into one clade. These findings are consistent with the phylogenetic position of B. gibsoni, B. duncani and B. orientalis based on the amino acid sequences of cox1 and cytb [26, 27, 30].
{kind=link}