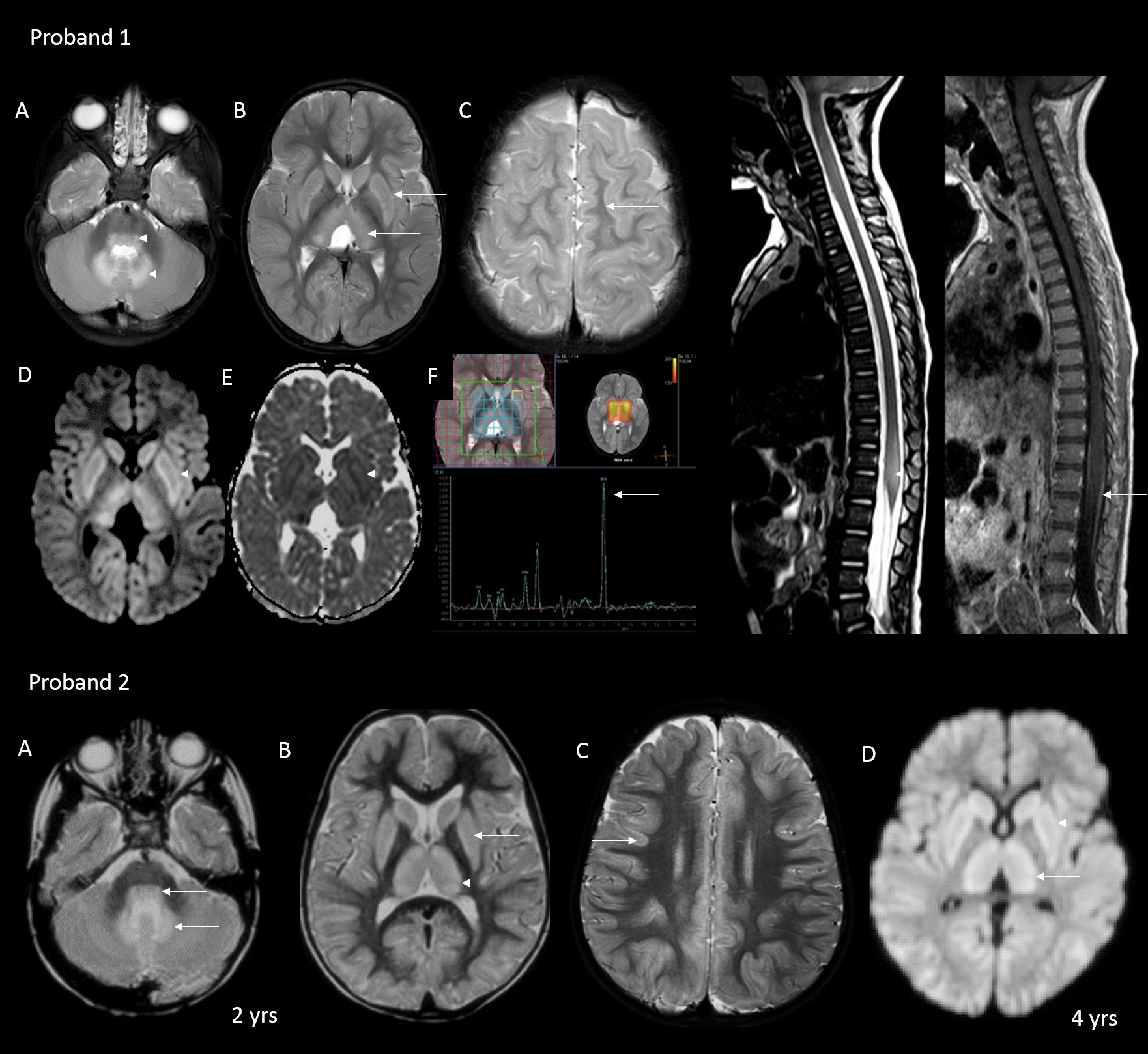
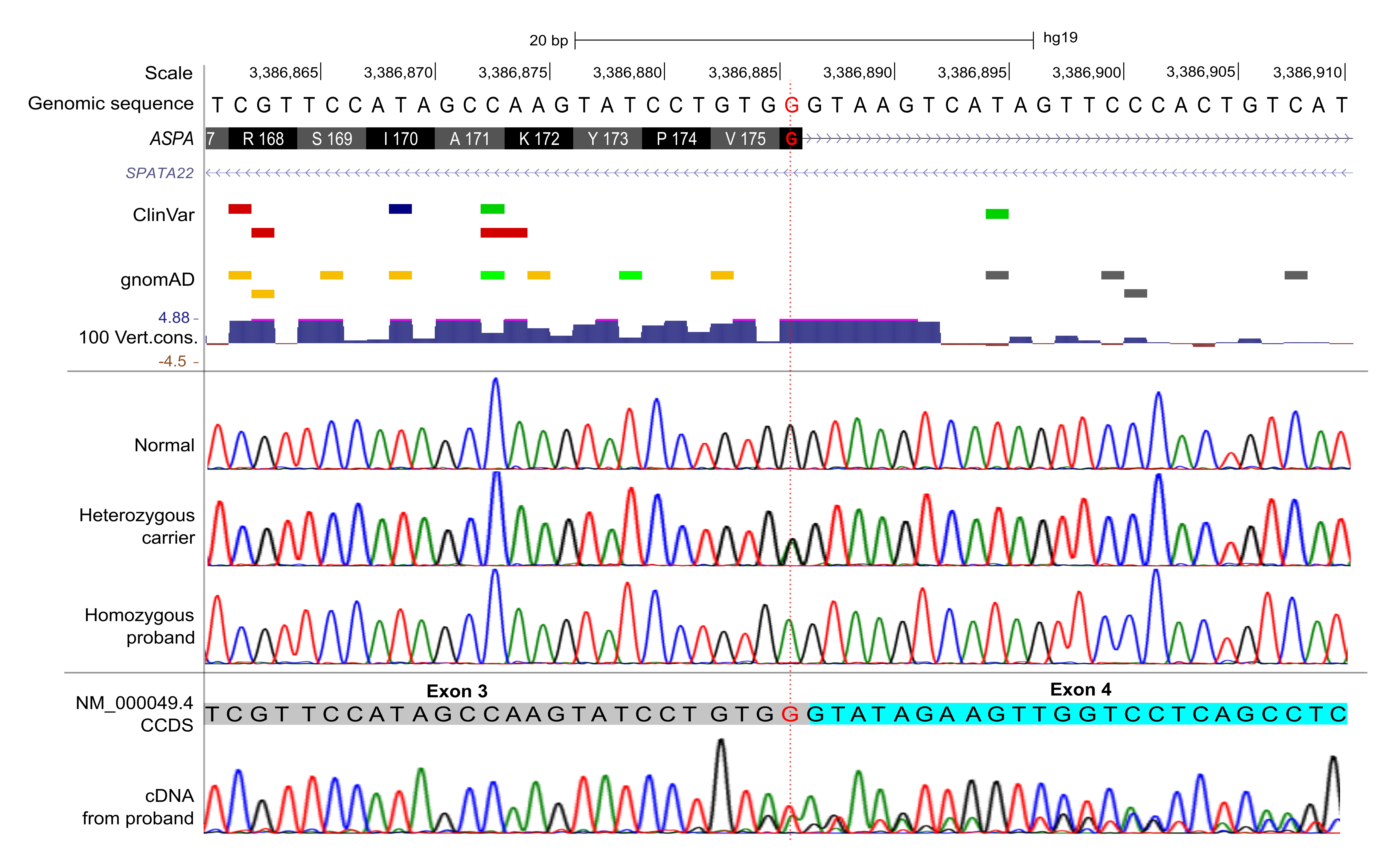
Both the probands harbored a novel homozygous variant c.526G>A (chr17:3386886G>A;GRCh37) in exon 3 also confirmed by WES. The missense variant ASPA(NM_000049.4):c.526G>A (p.Gly176Ser) has not been reported previously in ClinVar/HGMD or in the population databases such as gnomAD, 1kG, GenomeAsia100k, and our in-house database. The variant is predicted to be damaging by both SIFT and PolyPhen2. The nucleotide c.526 is predicted to be highly conserved by GERP++ and PhyloP across 100 vertebrates. MaxEntScan [27] and NNSplice [28] revealed a splicing defect. varSEAK splice tool predicted the replacement of the consensus splice donor site by the activation of a cryptic splice donor site 4 bp upstream (new position; c.523G). The variant segregated in a Mendelian recessive fashion among the family members. Hence, this variant was classified as Pathogenic.
The cDNA sequencing revealed a frameshift pattern starting 1 bp upstream of the novel variant (Supplementary Fig. 2). Deconvoluting the cDNA sequence revealed possible presence of 2 transcripts. One transcript with the activation of the upstream cryptic splice site GT (at position c.523_524) as predicted along with splicing of the 4th exon. This would result in a premature termination at codon 176 (p.Gly176Ter) and theoretically many more premature termination codons further downstream. Such an mRNA is expected to undergo nonsense mediated decay (NMD) as the variant is 223 bp upstream of the last exon-junction complex (EJC). Second transcript with the retainment of canonical splice site (escaping the splicing aberration) resulting in a complete transcript. Probably this results in a normal length protein harboring the missense change p.Gly176Ser. This non-classical splicing defect due to an exonic mutation has been classified as type IV splicing defect [29]. Such examples have been reported previously [27, 30, 31]. This is in line with the reported literature that, a milder missense variant and another pathogenic LOF variant often leads to M/JCD. Although further studies in the form of NMD analysis/enzyme activity levels are required to confirm the above possibilities.
Fixation index
The history of migration of the TDC from their historical origin centuries ago probably with a bottleneck and a history of endogamy leads one to speculate possibly low genetic diversity. Fixation index (F) is the most basic measure of such a variation and the results were evidently concrete. On an average there was 0.5 to 0.7% reduction in heterozygosity in the TDC as compared to the expected estimates under random mating. We also noticed a higher median F value compared to individuals (with no history of consanguinity) inhabiting the same geographical location (Table 1). The deviation however, is an order of magnitude less compared to the individuals born of consanguineous parentage. This was an early indication to the possible role of endogamy but with a lesser degree than consanguinity in the TDC. Reduction in heterozygosity also implies an increased proportion of loci which are homozygous: the Wahlund effect [32].
Relationship of ROH frequency and size among populations
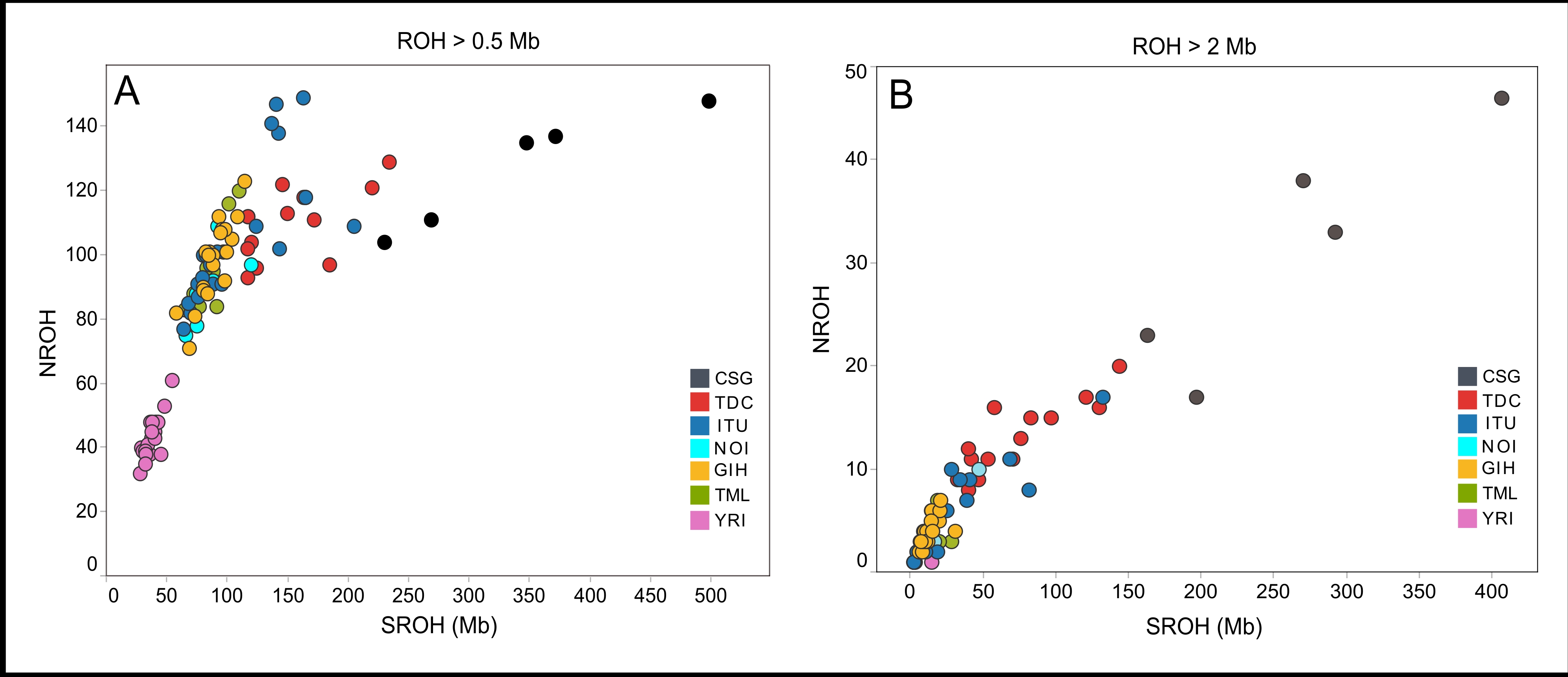
In Fig. 2A, the class I ROH represents similarity of the population living in the same geographical location as opposed to a population from a distant continent. For the class I ROH, no single subpopulation of Indian origin could be statistically differentiated based on the SROH and NROH. However, each of the subpopulation showed higher median SROH and NROH compared to YRI which were statistically significant (Supplementary Table S4). This is comparable to previously reported positive correlation of mean SROH with the increasing geographical distance of the study population from East Africa [12].
Size criterion for class II ROH was based on well documented literature evidence, where subpopulations begin to show differences in both mean SROH and NROH [10, 12]. It is clear from the data (Fig. 2B) that TDC had significantly higher SROH and NROH compared to other subpopulations with no history of parental relatedness (Supplementary Table S4). Even though small difference in the median values were found between TDC and individuals with parental relatedness (consanguinity), the difference was not statistically significant (P=0.117). These findings are concordant with the literature evidence of distribution of intermediate sized ROH [10, 11]. The intermediate sized ROH are often due to the effect of background relatedness determined by the effective population size [10, 11]. Historical evidence of probable population bottleneck in TDC and its current population size, might explain increased background relatedness and hence the above-mentioned findings. Finally, for the class III ROH (Fig. 2C), TDC showed higher SROH and NROH (P<0.005) compared to subpopulations with no history of parental relatedness. However, CSG differed significantly from TDC (P<0.0016), which is in line with the increased occurrence of multiple long ROH in individuals with recent parental relatedness. It is apparent from the data that, despite the absence of consanguinity, there is substantially higher than expected levels of intermediate and long sized ROH in the TDC. As can be noticed in Fig. 2D the TDC is on the left of the trendline suggesting that NROH is proportionately more than SROH. This is due to the fact that the class III ROH are few in TDC but the population is enriched with class II ROH. In contrast, the CSG group had the right shift because of numerous long class III ROH without a proportional increase in NROH. The pattern observed in TDC is analogous to worldwide patterns of numerous native populations with history of bottleneck, genetic isolation and with a small effective population size [11].
FROH is a better measure of autozygosity even in the absence of accurate and deep pedigrees, as FROH extracts true IBD as opposed F values from pedigrees (FPED) where founders are often poorly defined. FROH also reliably captures autozygosity originating from ancient as well as recent parental relatedness [11]. TDC had higher FROH values compared to all other subpopulations (p<0.05) except CSG (table 1). The overall results suggest an increased amount of autozygosity in the TDC, in spite of absence of recent parental relatedness.
IBD and kinship coefficients
Results of the kinship coefficient φ as per KING between pairs of individuals showed accurate relatedness among individuals with self-reported pedigree relationship. The results also unexpectedly unearthed cryptic relatedness with higher φ than expected among TDC individuals who were unrelated as per the pedigree (Supplementary Table S1). The propIBD estimation-based relationship inference provides more robust estimation of relatedness and the results of which were concordant with φ results. Refined IBD used a robust IBD segment caller showed that, the summated fraction of genome that is identical by descent (LOD > 3.0) in the TDC founders were several folds higher than unrelated individual pairs corroborating with the findings of KING estimates. This can be explained by the equation f=(1/2)i(1+Fa) [33]; where Fa is the cryptic probability of a founder in a pedigree who himself/herself is a product of inbreeding. Fa values increase in a population with endogamy alone or bottleneck superimposed with endogamy. Given the history of TDC, the inflated kinship coefficient/IBD estimates is not startling.
Haplotype analysis
As evident in Fig. 3, the family 2 harbored an unbroken segment of size 7.96 Mb which was segregating in the Mendelian fashion. The size exactly matches with the ROH found in the proband. However, in family 1 the haplotype has undergone reduction in size probably because of at least two independent ancestral recombination events, one each on the paternal and maternal side. The sizes of the segregating haplotypes were 4.73 Mb and 1.02 Mb respectively. The core haplotype of 1.02 Mb consisting of 147 SNPs is common to all the carriers and the affected. The Dʹ and r2 values between SNPs in the region of the haplotype were substantially high compared to what was found in other subpopulations. The sequence of the haplotype was also confirmed using PHASE 2.2 program. Further, the size similar to this haplotype was not found among any other individuals from various populations analyzed.
Population structure
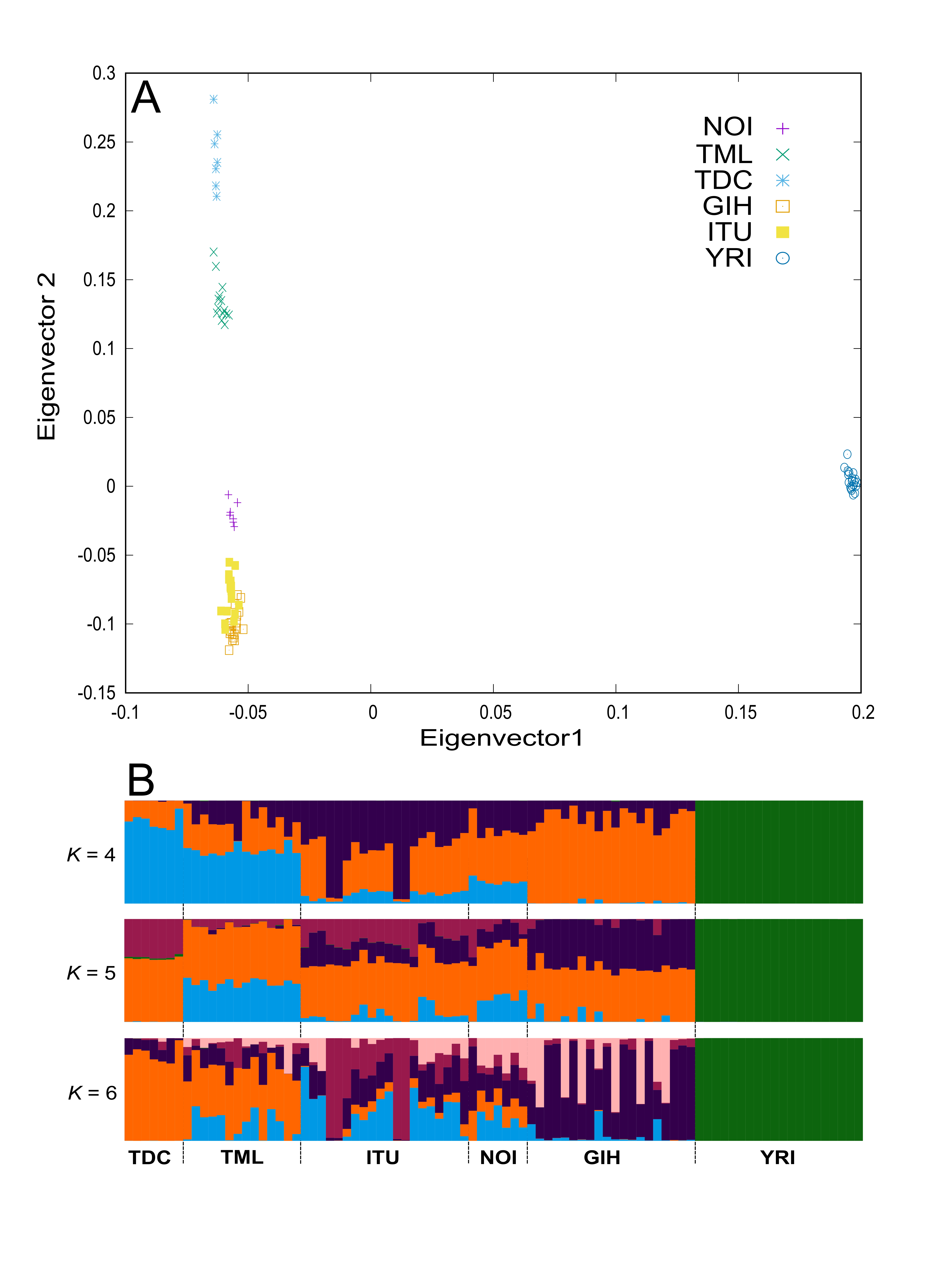
PCA clearly grouped individuals into distinct clusters which correlated well with their geographical location and native languages (Table 2). The data was in good agreement with eigenvalues, Tracy-Widom statistic, and ANOVA P-values (Supplementary Table S5). Clear cut population differentiation between TDC and TML who are inhabiting the same geographical region was noted (Fig. 4 A). Both the populations were far removed from the other subpopulations such as NOI, GIH and ITU along the PC1 and PC2 axes. Further, all these populations aligned on the PC2 axis and were at a notable distance from YRI on the PC1 axis as documented in previous studies [21]. This was the first clear indication as to the differentiation of TDC from other populations and presence of a population structure. FST output after correcting for inbreeding, as per the Balding-Nichols model (Table 3) showed moderately great differentiation of TDC from YRI and moderate from other subpopulations of the Indian ancestry without any overlap [34]. The only overlap was between ITU with GIH (FST = 0.003; threshold for overlap considered < 0.004) [35].
Treemix provided the conclusive maximum likelihood tree representing the topological relationship of the subpopulations. The Newick format ML tree (Fig. 4 B) shows the split of TDC and TML from a common ancestral population which in itself is a split from the common ancestors of ITU. The length of the branch representing TDC clearly shows the significant divergence (P=7x10-5) and probably minimal or no migration between TDC and TML. STRUCTURE results concluded the same (Supplementary Fig. 4).
There is enough evidence as per the results of FST, PCA, STRUCTURE and Treemix to support a process akin to ‘lineage sorting' having taken place. This is probably due to the significant population contraction (leading to drift) and long genetic isolation. As per Island model (Expected FST= 1-e-t/2Ne), a significant population contraction resulting in drift will increase differentiation and there by FST [36]. One of the main assumptions of the Island model is that no migration has taken place between the subpopulations. Based on self-report and the strict cultural practices of the TDC collating with the above-mentioned results, it is evident that the TDC have undergone genetic isolation with hardly any migration.
Mutation dating
Assuming a correlated genealogy, the mutation arose 15.1 generations ago, with a confidence interval of 3.3 - 26.5 corresponding to around 375 years (assuming one generation = 25 y). This coincides well with the age of historical migration of the first founders of TDC to the current location of inhabitation [4]. Gamma method as opposed to coalescent based methods, in fact provides an estimation of age of the most recent common ancestor (MRCA) rather than the exact age of the mutational process.
{kind=link}
{kind=link}
{kind=link}
{kind=link}