Cell culture
RAW264.7 cells (a mouse macrophage cell line obtained from Cell Bank of Chinese Academy of Sciences, Shanghai, China) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Sigma Inc., St. Louis, MI, USA) containing 10% heat-inactivated fetal bovine serum (FBS; Gibco, Grand Island, NY, USA), 100 U/mL penicillin (Sigma Inc.), and 100 mg/mL streptomycin (Sigma Inc.) at 37°C in a humidified atmosphere with 5% CO2.
S. aureus infection
S. aureus (strain NCTC8325; a gift from Professor Sun, Department of Microbiology and Immunology, School of Life Sciences, University of Science and Technology of China, Hefei, China) proliferated overnight in 10 mL Luria–Bertani (LB) broth with kanamycin at 37°C. S. aureus was washed with chilled nonpyrogenic saline, resuspended in nonpyrogenic saline at an OD650 of 0.1, and then kept on ice until infection. S. aureus was diluted to reach a multiplicity of infection (MOI) of 10:1 (bacterium: cell) in DMEM for 1 h as in our previous study [15].
RNA interference
Specific small interfering RNAs targeting TLR2 were purchased from Shanghai GenePharma Corporation. TLR2 siRNA (sense: 5’-GGAACAGAGUGGCAACAGUTT-3’ and antisense: 5’-ACUGUUGCCACUCUGUUCCTT-3’) and Control siRNA (sense: 5’-UUCUCCGAACGUGUCACGUTT-3’ and antisense: 5’-ACGUGACACGUUCGGAGAATT-3’) were used. Adherent cells were transfected with siRNAs by applying Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) in accordance with the manufacturer’s instructions. Knockdown efficiency was determined by western blot analysis.
Immunocytochemistry
RAW264.7 cells (5 × 105 cells/well) were seeded into glass-bottomed dishes with 1 mL DMEM for 12 h and were washed three times with phosphate buffer saline (PBS) to remove nonadherent cells. After being transfected with TLR2 siRNA or treated with U1026 for 2 h as in the previous study [16], the cells were infected with S. aureus (MOI: 10) at 37°C for 1 h in antibody-free DMEM. The cells were then fixed in 4% paraformaldehyde, permeabilized with 0.3% Triton X-100 (Sigma-Aldrich) for 10 min, washed with PBS five times, and then blocked with 5% donkey serum for 30 min at room temperature to inhibit nonspecific immunoreactivity. Next, the cells were incubated overnight at 4°C with primary goat anti-mouse Gal-3 antibody at 1:100, rinsed three times in PBS, and incubated with FITC anti-goat secondary antibody (Jackson Immuno Research Lab, West Grove, PA, USA) at a 1:200 dilution. The cells were counterstained with DAPI (Sigma-Aldrich). Images were captured under a Zeiss LMS880 confocal microscope.
Animals
Female C57BL/6 mice (WT, 6-8 weeks, 20-25 g) were obtained from the Shanghai Laboratory Animal Center. TLR2 deficient (TLR2-/-) mice on C57BL/6 background were granted by Dr. ZG. Tian (Institute of Immunology, School of Life Sciences, University of Science and Technology of China). WT and TLR2-/- were housed under specific pathogen-free conditions at the Department of Laboratory Animals Center. All of the animal experiments were strictly conducted under protocols approved by the Institutional Animal Care and Use Committee of the Bengbu Medical College.
Experimental protocols and treatment
The protocol applied to establish OVA-induced allergic airway disease in mice was in accordance with a previous study [17]. Briefly, the mice were sensitized by intraperitoneal injection of 10 μg OVA (Chicken Egg OVA, Grade V; Sigma) and 1 mg aluminum potassium sulfate (Sangon Biotech, Shanghai, China) in 0.5 mL saline on day 0, then challenged for 30 min per day with 1% aerosolized OVA on days 14–20. Control mice were saline-sensitized and challenged with nebulized saline solution. On days 14–20, the WT and TLR2−/− mice from the U0126-treated group were pretreated with specific MAPK/ERK inhibitor U0126 (30 mg/kg, Sigma-Aldrich) or vehicle (polyethylene glycol (PEG), Sigma-Aldrich) intraperitoneally, diluted in 0.1 mL sterile saline 30 min prior to OVA challenge as described previously [5]. All of the mice were sacrificed after the last challenge; on the left lung, bronchoalveolar lavage and subsequent differential cell counting and enzyme-linked immunosorbent assay (ELISA) were performed, while the right lung was used for histopathological analysis and western blot.
Histological analysis
Lung tissues were fixed in 4% paraformaldehyde. After that the paraffin-embedded tissues were sectioned (5-μm thickness) and stained with hematoxylin/eosin (H&E) or periodic acid-Schiff (PAS) to measure inflammatory cellular infiltration and mucus productions, respectively. The quantitative analysis of peri-bronchial inflammation in H&E-stained lung slices was performed by determining the number of rings of inflammatory cells around bronchial, and the mucus productions quantification was accomplished by assessing the number of PAS+ cells in the airway by the perimeter of the basement membrane (Pbm) as our previous study [18].
Bronchoalveolar lavage fluid (BALF) and cellular analysis.
BALF was collected from the left lung with 0.5 mL of ice-cold PBS and was centrifuged at 700 g at 4°C for 5 min. After that the cell pellets harvested from BALF were resuspended in 200 μL PBS. Total cells were counted in a hemocytometer, and differential cell count was determined by staining cytospins of BALF samples with Wright Stain solution (Sigma). At least 200 cells were counted for each mouse. The supernatants were stored at -80℃for ELISA analysis.
ELISA
The secretion of IL-4, IL-5, and IL-13 in BALF and OVA-specific IgE in serum were measured by ELISA using specific kits from Cusabio (Wuhan, China). The levels of Gal-3 in BALF and serum were tested by ELISA kits from Bio-Swamp (Wuhan, China) in accordance with the protocols from the manufacturer. The sensitivity for IL-4, IL-5, IL-13, and Gal-3 was 0.39 pg/mL, 7.8 pg/mL, 7.8 pg/mL, and 0.078 ng/mL, respectively.
Western blot
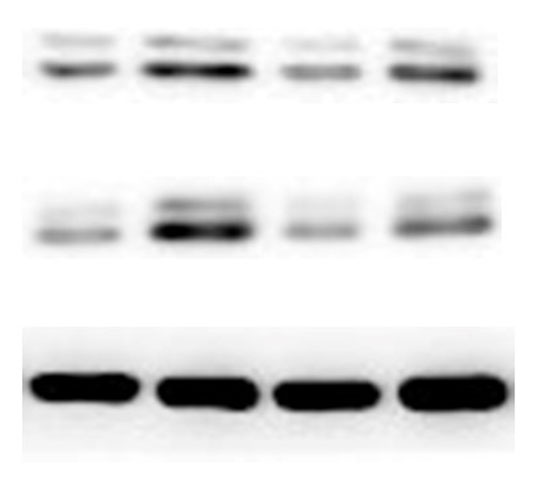
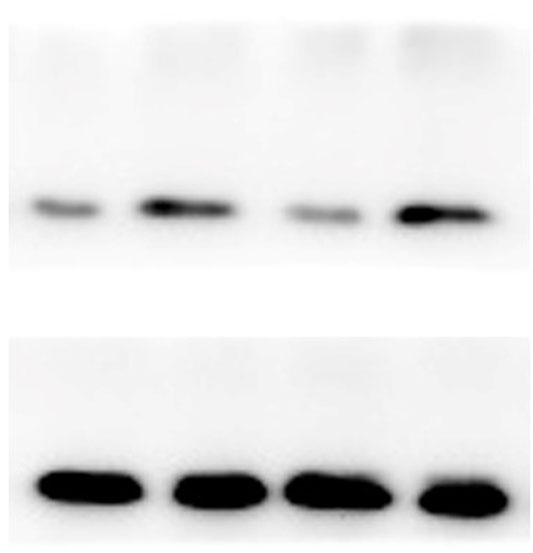
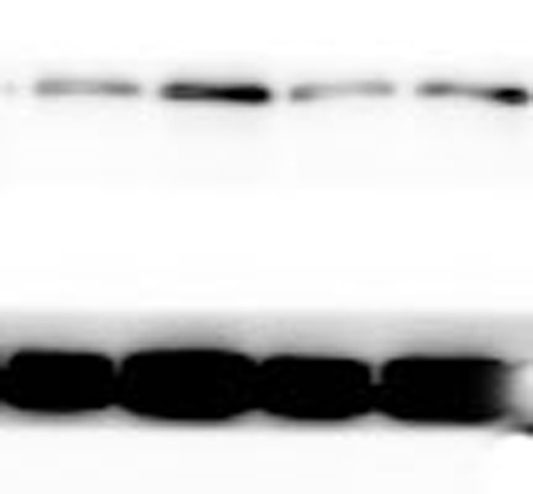
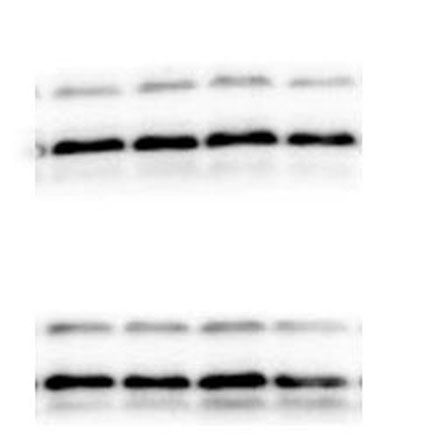
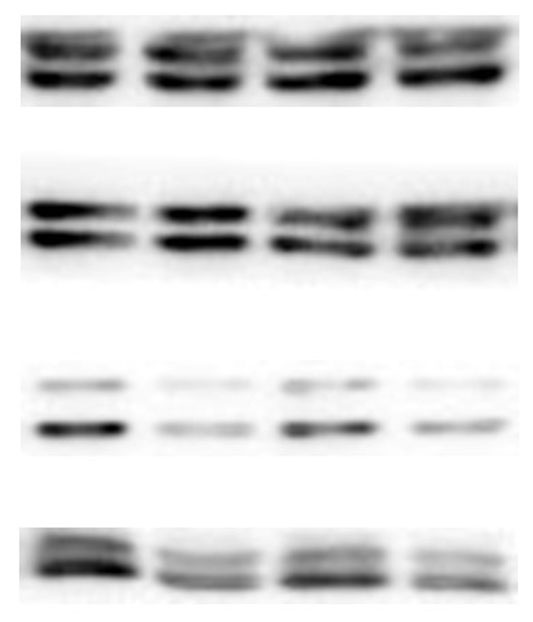
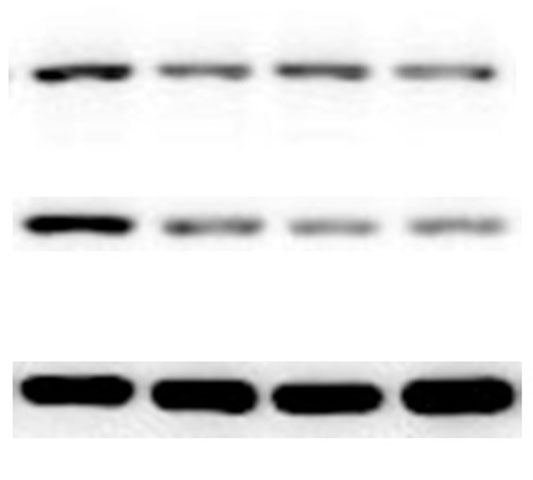
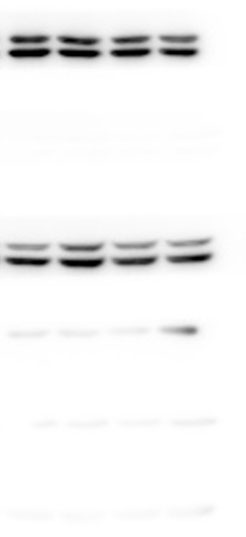
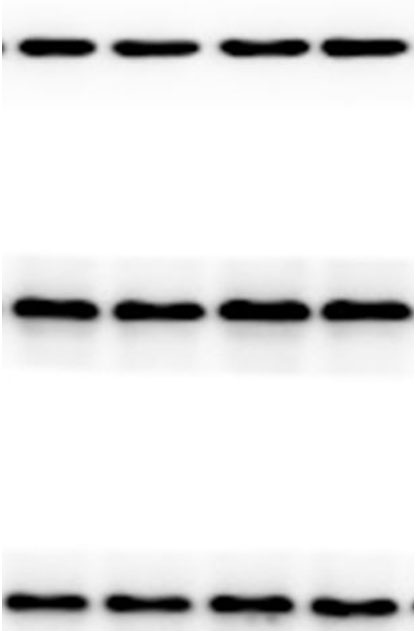
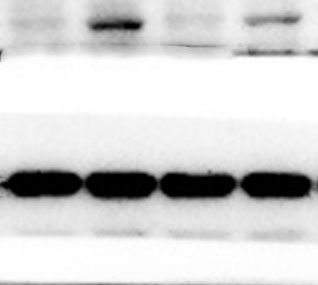
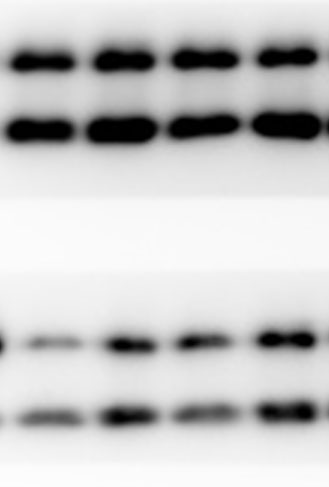
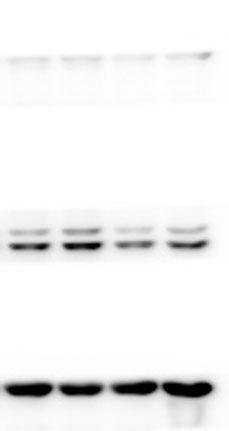
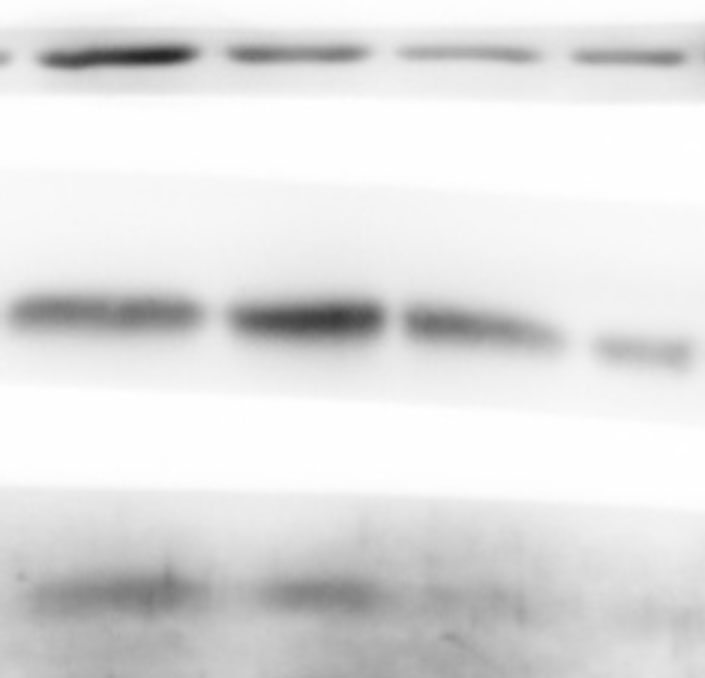
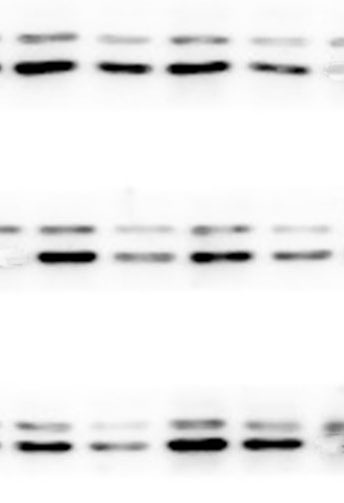
The lung tissues were homogenized using a homogenizer with RIPA buffer containing protease inhibitor cocktail (Roche, Indianapolis, IN, USA) and phosphatase inhibitor PhosSTOP (Roche). Total protein was separated by SDS-PAGE and transferred to PVDF membranes (Millipore, Billerica, MA, USA). The membranes were blocked in 5% nonfat milk and then incubated with primary antibodies. including anti-p-ERK/ERK (1:1000), anti-p-p38/p38, anti-p-JNK/JNK, and anti-TLR2 (1:1000) (Cell Signaling Technology Inc., Beverly, MA, USA), anti-galectin-3 (1:500) (Santa Cruz Biotechnology, CA, USA), and anti-GAPDH (1:2000) (KANGCHEN Biotech, Shanghai, China). The membranes were subsequently incubated with HRP-conjugated anti-rabbit IgG (1:2000) (Promega, Madison, WI, USA) and polyclonal rabbit anti-mouse IgG (Dako, Copenhagen, Denmark), and all blots were detected with enhanced chemiluminescence (ECL; Thermo Scientific). For quantitative analysis, intensity of protein bands was determined using ImageJ 1.38 software (NIH, Bethesda, MD, USA).
Statistical analysis
Statistical analysis was performed using SPSS 16.0. All data were expressed as the mean ± standard deviation of triplicate samples and were representative of at least three separate experiments. Independent-sample t test or one-way ANOVA with a post-hoc Bonferroni test was used for all statistical analyses. P values <0.05 were considered significant.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}