4.1. Study sample and image acquisition
A demographic summary of the samples used in this study is presented in Table 1. Analyzed imaging data included T1-weighted brain MRI of 307 individuals with ASD (25.4 ± 9.8 years, 273 males) and 362 age- and sex-matched TD controls (25.8 ± 8.9 years, 309 males) between the ages of 16 and 64 shared by the ABIDE (Autism Brain Imaging Data Exchange) consortium.24 Only data from 16 sites with more than 10 controls within the age range were included to ensure robust site-wise image harmonization, as outlined in 4.3. A summary of the 16 sites can be found in Supplementary Table 7.
For comparison, data from the PHENOM consortium that were used to derive neuroanatomical dimensions of SCZ21 were also investigated: 307 individuals with SCZ (30.9 ± 7.3 years, 199 males) and 364 TD controls (29.5 ± 7.0 years, 203 males). Additionally, 18,678 individuals (57.4 ± 4.3 years, 8,015 males) from the UK Biobank cohort25 representing the general population were investigated. A broad range of disease diagnoses based on the clinical history was also provided (https://biobank.ctsu.ox.ac.uk/crystal/coding.cgi?id=19&nl=1). All participants recruited in the above studies provided informed written consent according to the protocol approved by each of their Institutional Review Boards.
4.2. Image preprocessing and quality assurance
All raw T1-weighted MRI (182×218×182 voxels) from the ABIDE (N = 778 individuals initially) were manually examined for head motion, image artifacts, or restricted field-of-view. Two individuals were excluded due to significant noise in the images. Images were then corrected for magnetic field inhomogeneity, and a multi-atlas segmentation58 was applied to obtain brain volumes in 145 anatomical ROIs (Supplementary Table 1). Two individuals were further excluded due to under-segmentation. The final sample (N = 669) after matching the two groups for mean age and sex has been outlined above. The MR images of PHENOM and UK Biobank followed the same quality assurance procedure and were detailed elsewhere.21,36
4.3. Image harmonization
Site-wise differences in the 145 ROI volumes were linearly harmonized using ComBat59, while preserving effects of age, sex, and ICV, based on the control group of the ABIDE sample. For the control-based site-wise harmonization purposes, only cognitively normal UK Biobank participants without the history of depression (N = 1,510), diabetes mellitus (N = 742), stroke (N = 123), bipolar disorder (N = 60), or any mental, behavioral, neurodevelopmental disorders (code F01-F99 from the ICD-10;26 N = 1,554) were treated as the reference control group (N = 15,325). Subsequently, the ROI volumes were linearly adjusted for age- and sex-wise differences, based on the trends in the TD group. For every validation or test dataset (PHENOM or UK Biobank), to prevent information leakage, its harmonization was performed separately and based on the TD group of the training dataset (on ABIDE). Then the corrected ROIs were normalized to z-scores and were used as features in the HYDRA models. Supplementary Fig. 11 contains a flowchart to summarize the harmonization procedure. The efficacy of the harmonization was manually checked post hoc, and the final dimension scores of the TD controls showed insignificant site-wise variations (P > 0.9, one-way ANOVA) (Supplementary Fig. 12)
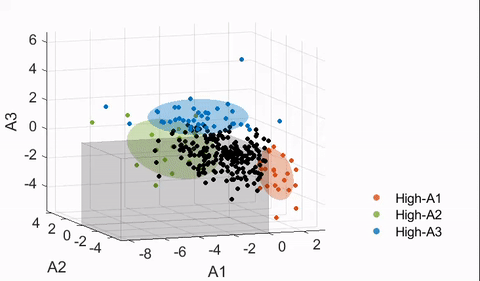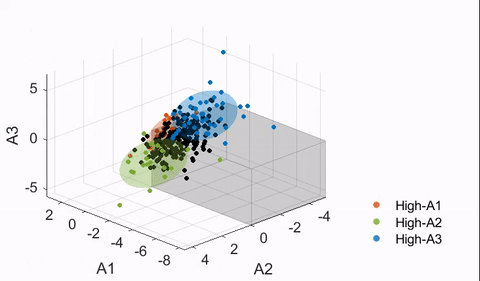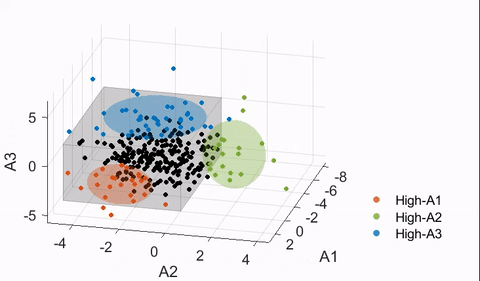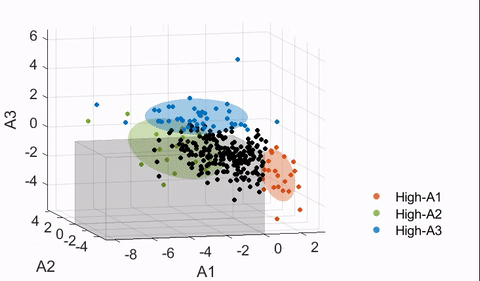
4.4. Finding neuroanatomical dimensions with HYDRA
Details of the HYDRA model can be found in Varol et al.17 In essence, a HYDRA model attempts to find a pre-defined number of linear support vector machine (SVM) hyperplanes (i.e., the polytope) that together enclose the control distribution while maximally separating the control group from the affected group (ASD in this case). Then, each hyperplane can be considered as a plane that is orthogonal to a dimension in which the affected group deviates from the control distribution, and projections of the data points onto this dimension can be quantified (denoted here as a “dimension score”).
The HYDRA model implemented in MATLAB R2020a was trained with the following default parameters: 10-fold nested cross-validation, initialization with determinantal point processes (DPP), L1 regularization, C = 0.25 (regularization parameter). The number of dimensions/clusters (k) was varied between two and eight to find the optimal dimensional space to capture the heterogeneity of ASD neuroanatomy. As the estimated solution may differ depending on the initialization, each nested model was trained 50 times for the main model, and the derived dimension scores were averaged.
To evaluate the stability of the trained models, a metric known as the adjusted Rand index (ARI)27 was used. ARI is a measure of agreement between partitions and is frequently used in validation of clustering analyses for its relative insensitivity to the number of clusters. ARI was computed per HYDRA model by treating the neuroanatomical dimensions as clusters (i.e., a subject is assigned to a cluster corresponding to the dimension with the largest score received). To test the significance of each ARI measure, a permutation test was followed with the group labels (ASD versus TD) shuffled. This was repeated 100 times to build the “null distribution” of the ARI, and t-tests were performed to compute significance. Both a high ARI and a significant difference from the null distribution were desired in a stable HYDRA model.
A set of thorough cross-validations were also performed. First, leave-one-site-out cross-validation was conducted for the three largest sites. Second, all female participants (N = 87) were held out during the training and used in the testing. Third, a stratified 10-fold cross-validation was performed with stratification by site, sex, and diagnosis. Fourth, a stratified 2-fold (split-half) cross-validation was performed for the model with the optimal k to verify that the neuroanatomical patterns captured by the dimensions were reproducible. Each cross-validated model was trained 20 times with different random initialization. Pairwise mean absolute error (MAE) between dimension scores was computed.
The ASD dimension scores from the stratified 10-fold cross-validation were used as the final scores for the individuals of ABIDE. The final model that was trained with the full ABIDE dataset was applied to compute the ASD dimension scores for the PHENOM and UK Biobank participants. The raw dimension scores were adjusted for ICV using association parameters estimated in the control group. Both sets of the raw and the ICV-corrected dimension scores were investigated.
4.5. Characterizing anatomical patterns of ASD dimensions
The T1-weighted images were transformed to the voxel-based RAVENS (Regional Analysis of Volumes Examined in Normalized Space) maps60 for gray and white matter and were harmonized using the same methods as with the ROIs. Pearson correlations between the final ASD dimension scores and the harmonized RAVENS maps were computed and the P-values were adjusted with the Bonferroni correction. Additionally, the dimension scores were correlated to the brain age gap (predicted age based on neuroanatomy minus the actual chronological age)30 for the individuals of UK Biobank.
4.6. Associations to clinical variables and other diagnoses
ASD dimension scores were correlated with the ADOS scores29 and three types of IQ (full-scale, verbal, and performance). The majority of the ABIDE participants (> 82%) received variations of the Wechsler IQ tests.28 The correlation was computed first with all IQ scores, and then with only scores from the Wechsler tests to confirm whether the same trends were observed (Supplementary Table 3). The dimension scores were also related to the medication status for individuals with ASD. Out of 203 individuals who reported their medication status, 61 (30.1%) were currently taking related medication (within three months from the MRI scan). Supplementary Table 4 provides the complete list of medications. A complete list of demographic and clinical variables investigated, including those from PHENOM and UK Biobank datasets, are summarized in Supplementary Table 2. All P-values were adjusted for multiple comparisons using the Benjamini-Hochberg false discovery rate correction when establishing significance.
4.7. Genetic quality check protocol
The genetic data for the UK Biobank participants (version 3) were downloaded from their website (https://www.ukbiobank.ac.uk/enable-your-research/about-our-data/genetic-data) on July 2021. We used the imputed genetic data in the current study. Details regarding the imputation can be found in the original UK Biobank paper.25 The genotyped and imputed SNPs (single nucleotide polymorphisms) from the UK Biobank were preprocessed with a standard quality check (QC) protocol (https://www.cbica.upenn.edu/bridgeport/data/pdf/BIGS_genetic_protocol.pdf).36
First, we extracted and excluded related individuals (2nd-degree related individuals) in the complete UK Biobank sample (N = 488,377) via King software relationship inference.61 Duplicated variants from all 22 autosomal chromosomes were then removed. Subsequently, we consolidated datasets for participants with both imaging and genetics data to have both modalities. Participants were removed if they had unmatched sex identities between genetically-identified sex and self-acknowledged sex. Lastly, subjects with i) more than 3% of missing genotypes, ii) variants with a minor allele frequency of less than 1%, iii) variants with larger than 3% of the missing genotyping rate, or iv) variants that failed the Hardy-Weinberg test at 1×10− 10 were removed. Finally, 33,541 participants and 8,469,833 SNPs passed the QC procedures to be used for the subsequent genome analyses. To adjust for population stratification,62 we derived the first 40 genetic principal components (PC) using the FlashPCA software.
4.8. Genome-wide associations
After the QC, 14,786 participants with European ancestry and within the age range of the ABIDE sample were filtered. The three ASD dimension scores and two SCZ dimension scores were treated as phenotypes of interest and were fit into multiple linear regression models for GWAS using Plink 2 (v2.0.0).63 Age (at the time of scan), age-squared, sex, age-sex interaction, age-squared-sex interaction, ICV, and the first 40 genetic principal components (PC) were used as covariates. The FUMA online platform (v1.3.7)34 was then used to annotate the genomic risk loci, lead SNPs, candidate SNPs, and independent significant SNPs (Supplementary Notes 1). For the lead SNPs identified in our GWAS, we then manually queried them in the GWAS Catalog.32 Lead SNPs that had not been associated with any clinical traits in the GWAS Catalog were labeled as novel. GCTA-GREML64 was used to estimate the SNP-based heritability of the dimension scores in the general population, while the same confounders used in GWAS were adjusted for. One-sided likelihood ratio tests were performed to derive the P-values.
4.9. Gene-level associations and gene set pathway enrichment
MAGMA (v1.0.8)33 was used for gene-level associations. Gene annotation was performed to map the valid SNPs (reference variant location from the Phase 3 of 1,000 Genomes for European ancestry) to genes (human genome build 37). The gene-level associations used the SNP-level GWAS summary statistics to obtain gene-level P-values between each phenotype and the 18,902 protein-encoding genes. In addition, gene set enrichment analyses were conducted using gene sets from the MsigDB database (v6.2),65 including 4,761 curated gene sets and 5,917 ontology gene sets. Bonferroni correction was performed for all tested genes (P < 2.64×10− 6) and gene sets (P < 4.68×10− 6). All other parameters were set by default in FUMA.
4.10. Gene expressions
FUMA provides a core functionality (GENE2FUNC) to analyze gene expression in the phenotype of interest (therein A1, A2, and A3). In particular, GENE2FUNC took the prioritized, mapped genes (by default in FUMA) as input. The gene expression heat maps were generated using GTEx version 8 (54 tissue types and 30 general tissue types).66 The average expression per label (log2 transformed in each tissue type) was displayed on the corresponding heat maps. All other parameters were set by default in FUMA.
4.11. Genetic Correlations
LDSC35 was used to estimate the pairwise genetic correlation between each dimension score and seven neurodegenerative and neuropsychiatric diseases, including Alzheimer's disease (AD), attention-deficit/hyperactivity disorder (ADHD), ASD, bipolar disorder, major depressive disorder (MDD), SCZ, OCD (obsessive-compulsive disorder). The details of the inclusion criteria were described elsewhere.36 LDSC uses the GWAS summary statistics from our study and previous studies for the seven selected diseases to determine the proportion of variance that two clinical traits share due to genetic causes.
{kind=link}